NEJM重磅综述:表观遗传在癌症治疗中的应用
染色质是最早确定的癌症治疗靶点,早在20世纪70年代,科学家们就已经开始设计可以和DNA甲基化相关的改变染色质的促分化药物。癌症基因组测序发现了众多编码调节染色质蛋白质的基因突变,促进了人们对于染色质复杂性及其在肿瘤生成中作用的精准理解。近日,《新英格兰医学杂志》(NEJM)发表重磅综述,概况了表观遗传相关的获批药物及其临床疗效、目前在研的表观遗传药物以及表观遗传治疗领域面临的挑战。
表观遗传概述
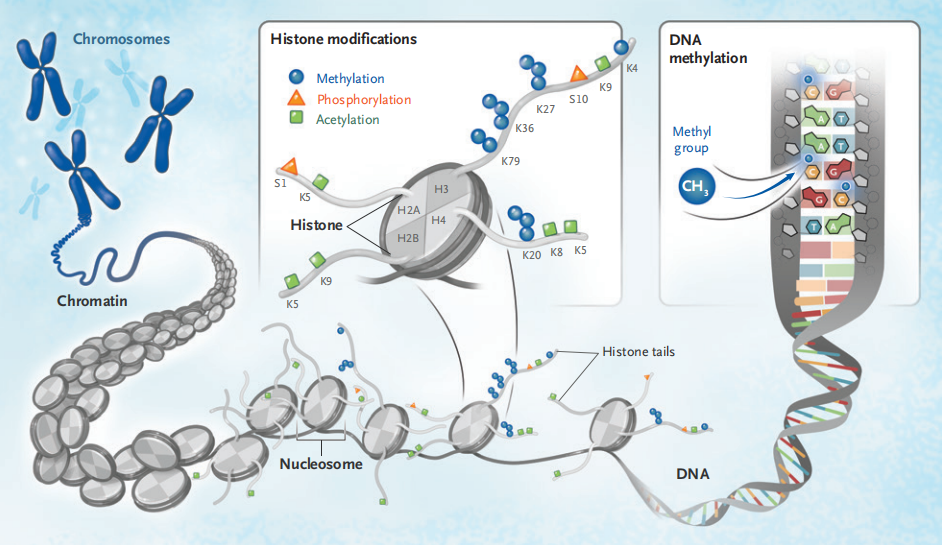
表观遗传始于DNA和组蛋白这两种在染色质结构和功能紧密相关的大分子物质。基础的染色质单位——核小体包括重复的146 bp的DNA片段,包裹于组蛋白八聚体的周围。组蛋白家族包括H2A,H2B,H3,H4和很多变异体,部分具有独特的功能。DNA和组蛋白通过调节亲和性和功能来进行修饰,这些修饰的改变是肿瘤的标志之一。表观遗传治疗致力于使DNA甲基化模式或促进持恶性表型的组蛋白翻译后修饰正常化。
调节DNA复制和修复以及RNA翻译的机制涉及调节DNA中胞嘧啶甲基化的酶,以及翻译后组蛋白修饰的多样化。富含赖氨酸的N端组氨酸“尾巴”的翻译后修饰包括乙酰化和甲基化,以及泛素化,磷酸化和类泛素化。尽管具有多样性,组蛋白修饰和它们的调节酶都十分特异,组蛋白修饰后,影响的残基在表观基因组中十分特异,因此科学家们发明了专用术语来描述这些修饰。例如,H3K9ac表明一个乙酰基集团(ac)加入了赖氨酸(K),位于组蛋白H3的第9个残基。H3K9me3表明3个甲基集团(me3)加入了上述氨基酸。乙酰化和甲基化通常是组蛋白的标志,而不是翻译后的修饰。乙酰化导致开放活跃的染色质状态,而甲基化更为复杂,其具有多种作用且取决于被修饰的残基。在某些位点如H3K9,甲基化和抑制的染色质状态有关;而其他位点例如H3K4,甲基化和基因转录有关。
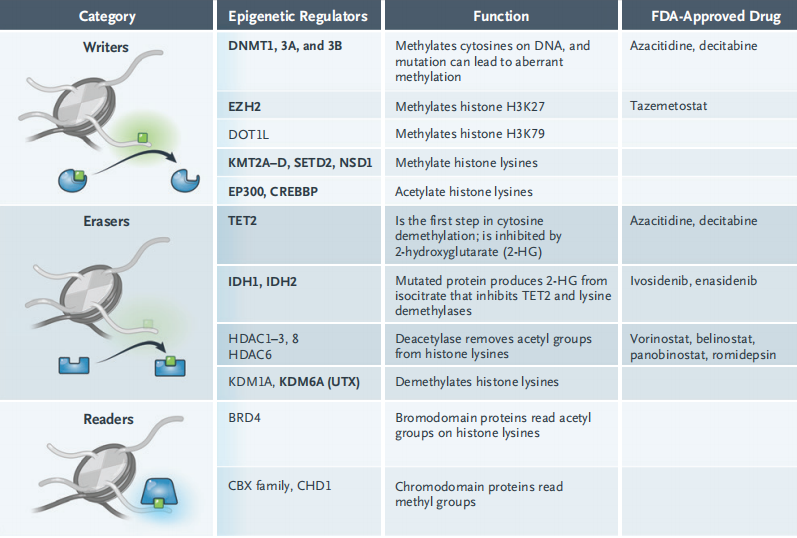
调节组蛋白翻译后表观遗传修饰的酶可以分为写入者、擦除者、读取者和搬运工,以区分超过700种调节染色质功能的蛋白质的作用。写入者负责加入修饰,包括DNA甲基转移酶(DNMTs)、组蛋白赖氨酸甲基转移酶(KMTs)和组蛋白乙酰转移酶(HATs)。擦除者包括组蛋白赖氨酸去甲基酶(KDMs)和组蛋白去乙酰酶(HDACs),移除翻译后修饰。结构域和染色质域蛋白可以“读取”乙酰化或甲基化的残基,因此它们被称为读取者。搬运工是染色质重塑蛋白,可以搬运染色体,允许基因转录。而名词塑形者和绝缘体则代表其他功能。
表观调节突变及其作为靶点的证据
随着千万癌基因组测序的完成,和每个表观遗传调节都存在突变的发现,我们对于染色体的复杂性及其在肿瘤生成中作用的理解日益增加。但是,单纯突变不一定具有功能性结果,没有功能就不是表观遗传治疗的靶点。一个蛋白质修饰突变可以产生癌基因驱动或干扰一个肿瘤抑制基因,但是也可以作为乘客突变不产生关键作用。随着时间发展,越来越多的突变被证实在癌症中具有重要作用。虽然很多表观遗传调节子的突变导致功能的获得或改变,但是大部分突变都破坏了构象或者产生了新的终止子,导致功能丢失。因此,表观遗传的关键是正确的时空出现正确的表观遗传调节子,而过多或过少的调节子都可以导致肿瘤生成。

多条途径导致导致DNA异常甲基化
多种异常DNA甲基化和肿瘤生成相关,具有异常甲基化的肿瘤中,启动子区域内或附近的CpG岛(DNA中具有高密度胞嘧啶-鸟嘌呤二核苷酸的区域)变为高度甲基化,而基因本身变得低甲基化。2010年,超过20%的急性髓系白血病(AML)的样本中都发现了DNA甲基转移酶写入者DNMT3A的突变,超过一半的突变位于R882氨基酸。但是,之后认识到R882突变主要是隐性的,导致催化活性降低,并导致局部低甲基化,而不是AML DNA野生型DNMT3A R882所见的超甲基化。DNMT3A的改变通常作为启动突变,可以导致祖克隆或基础前白血病克隆,而一旦累积更多的突变就可以导致明显的白血病亚克隆出现。祖克隆通过消退或复现而持续存在。
其他分子改变强调了DNA超甲基化的恶性潜能。一个例子是功能丢失突变,特别是TET2,在AML中十分常见。通过催化去甲基化的第一步——甲基胞嘧啶转变为羟甲基胞嘧啶——TET家族擦除者蛋白逆转了DNMT1 3A和3B催化的胞嘧啶甲基化,阻碍去甲基化,并导致异常DNA超甲基化。TET2改变在意义不明的克隆性造血(CHIP)中很常见。CHIP是一种发生改变的克隆,65岁以上人群发生率为10%,和血液肿瘤及冠状动脉疾病相关。
另一个例子是三羧酸(TCA)循环酶突变,非直接的表观遗传修饰导致DNA和组蛋白超甲基化。异柠檬酸脱氢酶1(IDH1)R132的突变和异柠檬酸脱氢酶2(IDH2)R172的突变改变酶活性,导致优先生成致癌物质2-羟戊二酸(2-HG)的异柠檬酸盐而不是&α;-酮戊二酸盐。TET功能丢失突变可以通过2-HG抑制TET酶活性,导致DNA超甲基化;同时,通过2-HG抑制赖氨酸去甲基酶导致组蛋白赖氨酸甲基化增加。琥珀酸盐和延胡索酸盐的累积发生于琥珀酸脱氢酶和延胡索酸脱氢酶功能丢失突变的过程与此相同。
DNMT3A,TET2,IDH1和IDH2突变在AML的发生率分别是28%,14%,9%和10%,类似的突变频率也发生在外周T细胞淋巴瘤(AILT)的血管免疫母细胞瘤亚型。实体瘤中也存在改变DNA甲基化状态的突变,但是R882突变很罕见。除了AML外,癌症基因组图谱(TCGA)泛肿瘤数据集中10767例样本中的DNMT3A突变率为2%,仅有2例突变发生于R882。TCGA泛肿瘤数据集中的TET2突变发生率为2%,包括实体瘤中由于蛋白质截断而造成功能丢失的亚群。TCGA泛癌症数据集中病源性的R132 IDH1突变发生率为4%,包括78%的低级别胶质瘤,14%的胆管癌,和4%的皮肤黑色素瘤。
异常DNA甲基化的治疗药物
在发现甲基化形式复杂性之前,DNMT抑制剂阿扎胞苷和地西他滨就已经开发和获批了。两个药物都是抗代谢药,整合入DNA后可以抑制DNMT活性,诱导低甲基化;阿扎胞苷同时还整合入RNA。16名骨髓异常增生综合征患者反应率为17%,20名AML患者的反应率是40%,美国批准了阿扎胞苷(2004年)和地西他滨(2006年)用于治疗骨髓异常增生综合征和慢性髓系单核细胞白血病。在欧盟,药物获批用于不适合行骨髓抑制的初治或AML患者的二线治疗。而单药用于实体瘤活性的证据则不那么强。
虽然TET2,IDH1和IDH2突变导致DNA超甲基化,DNMT抑制剂的活性还不像有效的靶向治疗所期待的那么好,而低甲基化是否介导反应还不清楚。此外,所有的表观遗传疗法活性都不限于携带突变的癌症,可能是由于DNA损伤等通用的机制促进了细胞死亡。
有两个药物可以靶向编码IDH1和IDH2的热点突变,已经被美国食品和药品监督管理局(FDA)快速通道优先审批作为孤儿药:enasidenib可以靶向IDH2,ivosidenib可以靶向IDH1,二者分别在2017年和2018年获批。这是作用于肿瘤代谢产物的第一种获批。两个药物都可以显著降低血2-HG水平。在第一个人体试验中,38.8%携带IDH2突变的AML患者对enasidenib有反应。使用ivosidenib或enasidenib治疗后,反应的患者可以观察到循环突变IDH1或IDH2等位基因的清除。以前发现只有完全缓解(CR)的患者可以延长生存,后来发现持续部分缓解(PR)的患者也可以获益,但是治疗后不到一年时间会出现耐药。耐药的出现至少部分是由于携带IDH1或IDH2突变的细胞只构成了白血病细胞的一部分而非全部。随着数据成熟,还是不清楚是否可以发现和克服实体瘤中活性的不足。虽然IDH1突变频率在胶质瘤患者中很高,此类患者中药物能否穿透血脑屏障十分关键。在一项进行中的1期研究中,vorasidenib具有很好的中枢穿透性,可以降低胶质瘤患者93%的2-HG水平。
异常的组蛋白甲基化
组蛋白尾巴赖氨酸的甲基化是异常的DNA甲基化,可以导致基因沉默,和肿瘤发生相关。几个KMT写入者可能和肿瘤生成相关,包括EZH2、NSD2、SETD2和DOT1L。滤泡淋巴瘤中存在EZH2的功能获得性突变。虽然Y641残基的热点突变降低了非甲基化的H3K27甲基转移酶活性,它们增加了H3K27me2甲基转移酶活性,导致抑制性三甲基化的H3K27me3和基因沉默。几个其他的功能获得性突变(如A677G和A687V)以及功能丢失性突变也是致癌性的,但是很多EZH2突变还不是完全清楚。KDMs功能丢失性突变也可以改变组蛋白甲基化状态。
KMT抑制剂
很多将EZH2作为药物靶点的研究正在进行。Tazemetostat现在是最成熟的EZH2抑制剂,其他正在进行临床试验的还有valemetostat,CPI-1205,和CPI-0209的2期研究,tazemetostat用于复发或难治的非霍奇金淋巴瘤患者,野生型和突变型滤泡淋巴瘤的反应率分别是35%和69%,反应持续时间分别为13个月和11个月。试验结果支持FDA在2020年6月批准tazemetostat用于滤泡性淋巴瘤。而具有热点突变的实体瘤中则研究结果寥寥。在10967名TCGA泛癌症样本中,大多数具有EZH2突变的实体瘤似乎都是功能丢失性突变。只有3例Y641热点突变,都是皮肤黑色素瘤,体外研究显示tazemetostat可能具有活性。
Tazemetostat其他方面的应用也获得了批准。活跃的基因表达需要核小体从DNA移出,这项任务由SMARC家族成员完成,SMARC家族成员是一种ATP依赖性酶,为SWI/SNF染色质重塑复合物的一部分。一些SWI/SNF家族蛋白质功能丢失性突变导致了缺乏拮抗的EZH2活性的失衡,增加了H3K27me3和基因沉默。抑制EZH2阻断了这种失衡,导致细胞死亡。体外研究中,几个SMARC家族成员,包括SMARCA2/SMARCA4、ARID1A和PBRM1等证实了这点,并在横纹肌肉瘤和上皮样肉瘤的SMARCB1/INI1缺失和功能丢失性突变中得到了临床验证,15%的客观反应率(ORR)使得FDA批准tazemetostat用于治疗上皮样肉瘤。同样,滑膜肉瘤中SS18-SSX病源性基因融合导致SMARCB1从SWI/SNF复合物中丢失,使得具有这种改变的细胞对tazemetostat敏感。因此,EZH2的活化、SMARC家族成员功能丢失性突变和SS18-SSX融合都影响H3K27甲基化状态,为多种改变导致相同表观遗传异常而促进肿瘤生成的另一个例子。需要行系统的检测确定是否SEI/SNF复合物的其他成员改变也可以导致对EZH2抑制剂的敏感性。
异常组蛋白乙酰化
组蛋白乙酰化水平被HDACs调节。HDACs可以清除乙酰基集团,并通过HAT写入者乙酰化组蛋白尾巴的赖氨酸。HDAC擦除者家族包括4大类酶,共11种HdACs集团:I,IIa,IIb和IV,都可被HDAC抑制剂抑制。组蛋白去乙酰酶实际上是一个不当的命名,除了I类酶定位于核内,HDACs也使胞浆内的蛋白质去乙酰化,因此叫做赖氨酸去乙酰酶更合适。
去乙酰酶过表达导致组蛋白乙酰化程度降低,染色质关闭,基因表达降低。HDACs很少发生突变,TCGA泛癌症数据集中体细胞突变发生率不足1%。神经内分泌肿瘤等中过表达更为常见。而HAT突变很常见。EP300和CREBBP突变常为功能丢失性突变,降低了组蛋白乙酰化,阻碍和转录因子底物的相互作用。
HDAC抑制剂
4种HDAC抑制剂——vorinostat,romdepsin,belinostat和panobinostat都获得了FDA批准,第5种chidamide(西达本胺)在中国接受了常规审批(目前已获批)。全乙酰化(如H3K9ac,H3K18ac,H3K23ac,H3K56ac,H4K5ac和H4K16ac)来自于HDAC抑制。通过直接结合HDACs,抑制剂阻止赖氨酸去乙酰化,允许不受限的HAT活性和超乙酰化。乙酰化中和了正电荷的赖氨酸,降低了其对负电荷DNA的静电吸引力,继而染色质松散开放可以进行抑制细胞生成或诱导分化表型的基因转录。这个过程是HDAC抑制剂的主要机制。但还不清楚转录基因选择是如何发生的,以及这到底是不是HDAC抑制剂导致细胞死亡的机制。HDAC抑制剂的作用机制可能更为复杂,涉及DNA和线粒体损伤。胞浆靶点的角色还不清楚,特别是靶向I类和II类酶的vorinostat,belinostat和panobinostat。由于II类HDACs移除胞浆蛋白质和组蛋白的乙酰基集团,II类抑制剂增加胞浆蛋白质乙酰化,促进转录因子如TP53和TBX5的活化,稳定细胞骨骼蛋白。和组蛋白乙酰化相比,总的胞浆蛋白质乙酰化改变的效应还不清楚。
Vorinostat和romidepsin被FDA批准用于治疗皮肤T细胞淋巴瘤,romidepsin和belinostat获批用于治疗外周T细胞淋巴瘤,panobinostat获批联合地塞米松治疗多发性骨髓瘤。批准vorinostat和romidepsin用于治疗皮肤T细胞淋巴瘤时,FDA不仅考虑到30%和34%的反应率,还考虑到患者通常具有疼痛而变形的皮肤症状。对于外周T细胞淋巴瘤,romidepsin和belinostat获得加速批准,反应率为四分之一到三分之一。Romidepsin的中位反应持续时间超过1年。
HDAC抑制剂在T细胞淋巴瘤的活性,以及具有AILT的患者DNMT3A,TET2和IDH2突变常见,支持一些T细胞淋巴瘤的表观遗传来源。因此,接受romidepsin和阿扎胞苷治疗的T细胞淋巴瘤患者达到73%的反应率。但是,在皮肤T细胞淋巴瘤中没有观察到AILT患者中DNA甲基化基因的再度突变。而没有携带突变的患者使用药物也有反应,这让人疑惑治疗反应到底是不是只来自于表观遗传。
研究中的表观药物
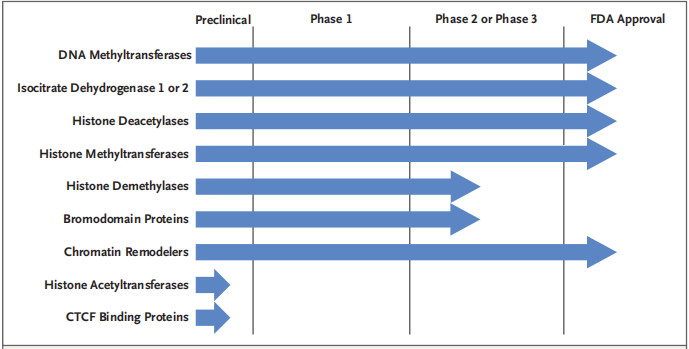
靶向写入者、擦除者、读取者和搬运者
美国现在有9种表观遗传药物可以用于标准治疗:2个DNMT抑制剂、4个HDAC抑制剂、2个IDH抑制剂以及最新的EZH2抑制剂tazemetostat。更多的写入者,擦除者和读取者抑制剂正在研究阶段。一种口服的DNMT抑制剂guadecitabine,正在以多种联合模式进行临床试验。其他KMT靶点抑制剂也还在研究中。MLL(混合系白血病基因,也就是KMT2A),menin(MLL支架蛋白)和DOT1L,唯一的H3K79甲基转移酶的相互作用也正在研究中。涉及MLL的基因重排可以产生和AF9,AF10和ENL的融合蛋白,之后将DOT1L招募到MLL靶点,改变表观遗传模式。目前相关研究已经设计了治疗策略来抑制DOT1L,MLL-menin相互作用,或者同时抑制二者,但是TOT1L抑制剂pinometostat单药的反应率很低,说明这种染色质重排非常复杂。VTP-50469的临床前活性表明MLL和menin支架蛋白的相互关系可以作为更核心的靶点,一个相关药物SNDX-5613的1/2期研究(ClinicalTrials.gov number,NCT04065399)正在进行。
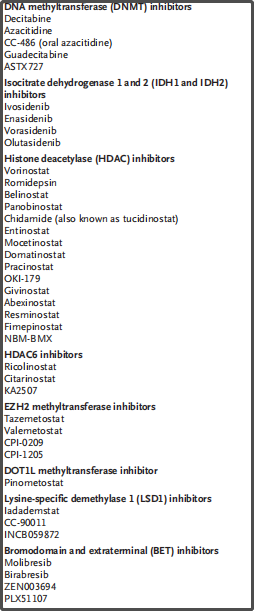
此外,组蛋白乙酰化的种类特异性HDAC抑制剂正在开发中,包括特异性的I类HDACs entinostat、HDAC8选择性抑制剂NBM-BMX(NCT03726294)以及HDAC6特异性抑制剂ricolinostat。具有CREBBP乙酰转移酶功能丢失性突变的细胞依赖残基p300乙酰转移酶活性,因此成为一种潜在的治疗策略。KDM1A(赖氨酸特异性组蛋白去甲基酶1[LSD1])擦除者靶向H3K4。H3K4me1或H3K4me2的甲基化是一个正常细胞生长和分化的关键活化标志。AML和骨髓异常增生综合征中LSD1的过表达降低了H3K4me1和H3K4me2,以及就像其他表观遗传异常,导致基因抑制。多种LSD1抑制剂正在开发中,包括iadademstat,可以导致白血病细胞分化以及偶尔引起AML反应。还有2个在研的LSD1抑制剂为INCB059872(NCT04061421)和CC-90011(NCT03850067),都在和其他药物合用。
溴区结构域和染色质结构修饰结构域读取者可以分别识别组蛋白乙酰化和甲基化位点。BRD4是溴区结构域以及额外终端域(BET)的成员,可以读取染色质的超乙酰化区域。BET抑制剂molibresib和birabresib在NUT(睾丸核蛋白)中线癌,一种罕见的携带特征致病性BRD4-NUT融合基因的癌症中的反应,证实了BRD4抑制剂的可行性。BET家族蛋白和很多参与癌基因过表达的转录复合物相关,因此多种BET抑制剂也在研究中。
靶向塑形者和绝缘体
针对塑形者和绝缘体等更新的靶点药物正在开发中。组蛋白中调节染色质的核小体和蛋白质复合物的塑形者,可以获得突变类似于或改变翻译后修饰所调节的蛋白质-蛋白质相互作用。组蛋白H3.3是表观遗传药物开发的靶点。90%的软骨母细胞瘤和20%的儿童胶质母细胞瘤在组蛋白H3.3的36位赖氨酸位置有一个蛋氨酸,它抑制了H3K36甲基转移酶,MMSET和SETD2,降低H3K36甲基化,改变了基因表达。
最后,一个新的表观遗传调节子最近被发现在癌症中也很重要。绝缘体是阻挡一个DNA区域和其他区域的DNA-蛋白质复合物,防止不正确的启动子-增强子相互作用和染色质沉默的播散。关于这些蛋白质作用的研究是在三维基因组结构上进行的。绝缘体蛋白CTCF结合特异性DNA序列,在正常细胞中创造一个相互作用的序列区域。癌症中异常的基因表达可以来源于CTCF的异常,DNA序列的异常,或相关蛋白如STAG2,RAD21或CHD8。泛癌症数据集中总的CTCF突变率为2%,在半数病例中突变具有致癌作用。
联合应用表观遗传治疗
表观遗传药物和经典化疗、靶向药物、其他表观遗传药物以及免疫检查点抑制剂(ICI)联合使用以增加血液肿瘤患者的反应率,并将应用范围扩展到实体瘤。虽然体外研究证实合用具有协同作用,但是效应依赖于环境,而临床结果通常令人失望。至今,只有panobinostat,硼替佐米和地塞米松接受了FDA加速审批。随着精准医疗策略的发展,希望对于表观遗传更深入的理解可能最终可以指导药物开发和联合应用。早在1983年,就发现DNMT-HDAC抑制剂联合具有协同作用,但是在AML的临床研究中没有达到一致的结果。涉及不同方案,疾病,新靶点和不同药物的研究可能可以避免早期研究的缺陷。考虑到它的长半衰期,entinostat可能格外适合联合治疗。最后,很多DNMT,HDAC以及EZH2抑制剂和ICIs联合的研究正在进行,诱导编码直接涉及免疫反应和诱导睾丸抗原或内源性逆转录病毒序列的免疫源性肿瘤的基因表达。
经验和展望
2020年,尽管9种表观遗传药物获批,表观遗传治疗进展还是主要集中在血液肿瘤,实体瘤还需要更多的工作。表观遗传学诞生已经接近50年,它的重要性,冗余性,关联性和脆弱性逐渐变得清晰;尽管已经填补了一些知识空白,而更多的欠缺日渐明显。我们根据特征对癌症的定义意味着只认识到了一套有限的特征,在表观遗传中也是如此。例如异常DNA甲基化已经作为反复出现的致癌事件,最少3种不同的通路可以导致表型改变:编码DNMT,TET和TCA循环某些酶的突变。这强调了需要发现其分子基础来靶向起作用的基因异常而不只是关注DNA甲基化的表型。举个例子,尽管DNA甲基化增加,DNMT抑制剂仅仅在TET和IDH突变中具有有限的价值。此外,虽然表观基因组的正常化被认为是一种治疗策略,还没有证实这可以用于治疗。
对于精准医疗的认识是将来开发表观遗传药物的关键。对于数百个可能的靶点,表观遗传改变是癌症中最常见的遗传。它们可能和基础突变出现得一样早,或者晚一些来驱动克隆亚组。明确这些异常的相互作用有助于帮助我们明确哪些突变导致癌症易感性。但是环境很重要,一种环境下的突变可能在另一种环境下就不再具有相同作用。在AML中,IDH1抑制就是一个典范。在胆管癌中,ivosidenib反应率很低,而14个月的总生存(OS)导致一些关键随机研究的开展。细胞类型也很重要:虽然表观遗传改变通常是残基特异性的,它们可能也可以影响数百个基因的表达,发生于正常细胞和癌细胞的不同细胞的不同的场景中。因此,相应的表观遗传净改变不可避免地依赖于环境和细胞。
另一个挑战来自于不像其他癌基因可以使用抑制剂,大多数表观基因改变为功能丢失性突变,很难治疗。设计可以干扰适用性机制的药物成为越来越多被使用的方法。FDA批准tazemetostat是基于无限制的EZH2活性和由于SWI/SNF复合物的SMARCB1的丢失而对tazemetostat有反应。相同的策略可能很快会应用于乙酰转移酶的功能丢失性突变。
虽然基础突变被驱动癌基因抑制后,表观遗传治疗可能不那么有效,但是对于癌症的早期发展阶段或较少获得性突变的癌症可能成功。对于胚系表观遗传异常,如发生于琥珀酸脱氢酶和其他TCA循环的酶,治疗可以既是预防性又是治疗性。
最后,包括DNMT,HDAC和EZH2抑制剂的表观遗传治疗,有时对于突变和野生型基因型都有效。应该集中于对于表观遗传机制更深入的理解以发展更好的治疗策略,而不是仅聚焦于扩展已有的适应证。
总 结
染色质依旧是重要的治疗靶点。已有的和正在研究的表观遗传治疗活性提供了这个策略有效的证据。考虑到潜在靶点数目之多,需要系统的发现和验证潜在药物靶点的方法来聚焦于药物研发,达到理想疗效。
参考文献
Bates SE. Epigenetic Therapies for Cancer. N Engl J Med. 2020 Aug 13
