MET THE UNMET NEEDS:MET通路异常及临床治疗概述
MET异常介导的致癌机制最早在人骨肉瘤细胞系中发现,自此不同癌种的MET信号通路异常也被发掘并被应用于指导临床治疗和相关靶向药物的研发。但在相当长一段时间内,MET变异并未有较好临床治疗策略,Crizotinib作为最早应用于MET扩增的肿瘤靶向药,尽管具有一定疗效但并非特异性针对MET通路靶向药物。近年来多项针对不同MET靶向药物研究再一次将MET变异推向风口浪尖,近日,《肿瘤探索》(Cancer Discovery)发表了一篇对于肿瘤MET异常小型综述,系统性阐述MET变异不同形式,临床治疗策略及耐药机制[1]。
MET通路作用机制及变异形式
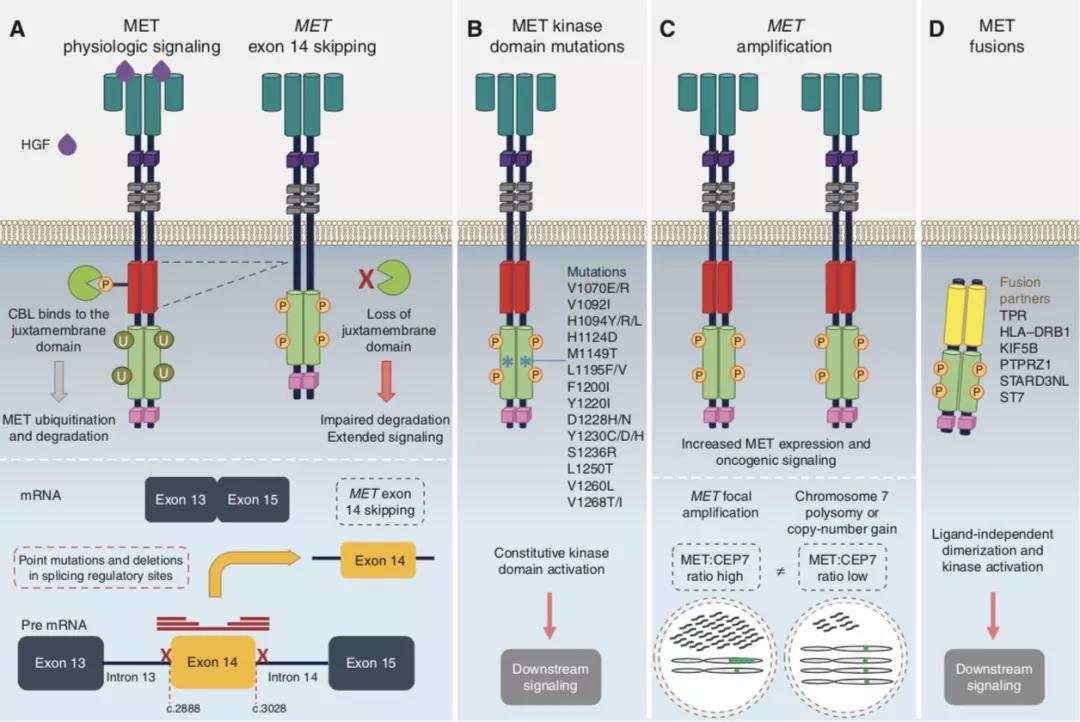
A. MET 14外显子跳读: 正常状态下MET信号通路传导,外源性肝细胞生长因子(HGF)与MET胞膜外受体段结合后诱导受体胞内段二聚化及磷酸化并活化下游多个信号通路,CBL蛋白结合到Y1003近膜域残基端(由14外显子编码),进而导致相应受体泛素化和降解。而位于剪接位点的MET 14外显子区点突变或缺失会导致外显子跳读,近膜域残基端缺失以及降低MET基因代谢并持续活化下游致癌信号通路;
B. MET激酶区突变:激酶区点突变导致MET非配体依赖性激活,进而持续活化下游信号通路;
C. MET扩增:MET扩增导致过度MET转录及表达,MET扩增介导的MET高表达会进一步增强下游致癌信号通路。
D. MET融合:MET激酶区重排所产生融合蛋白会二聚化为非配体依赖的形式并导致激酶区磷酸化从而异常活化MET信号通路。
MET通路激活机制
MET 14外显子变异
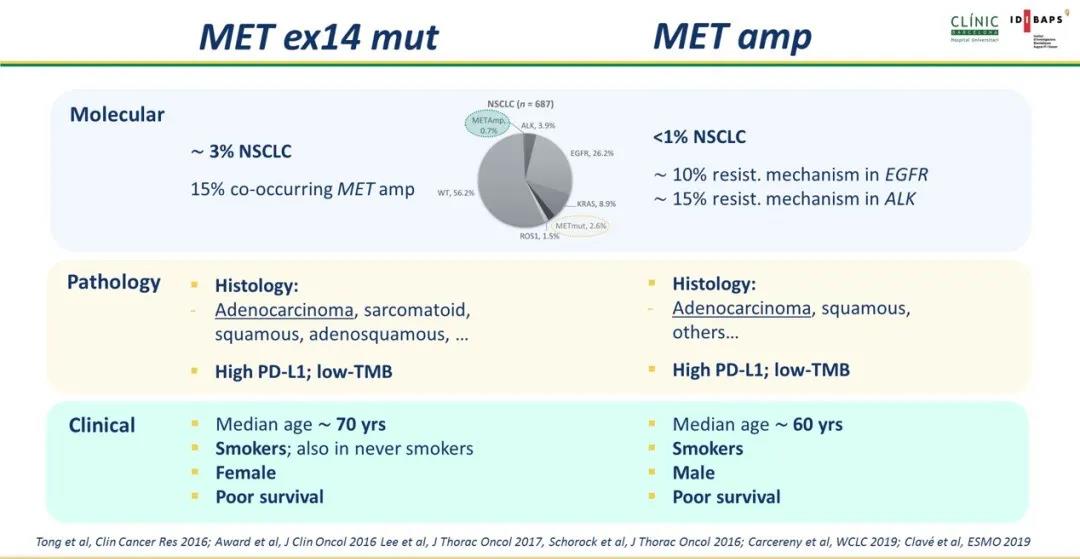
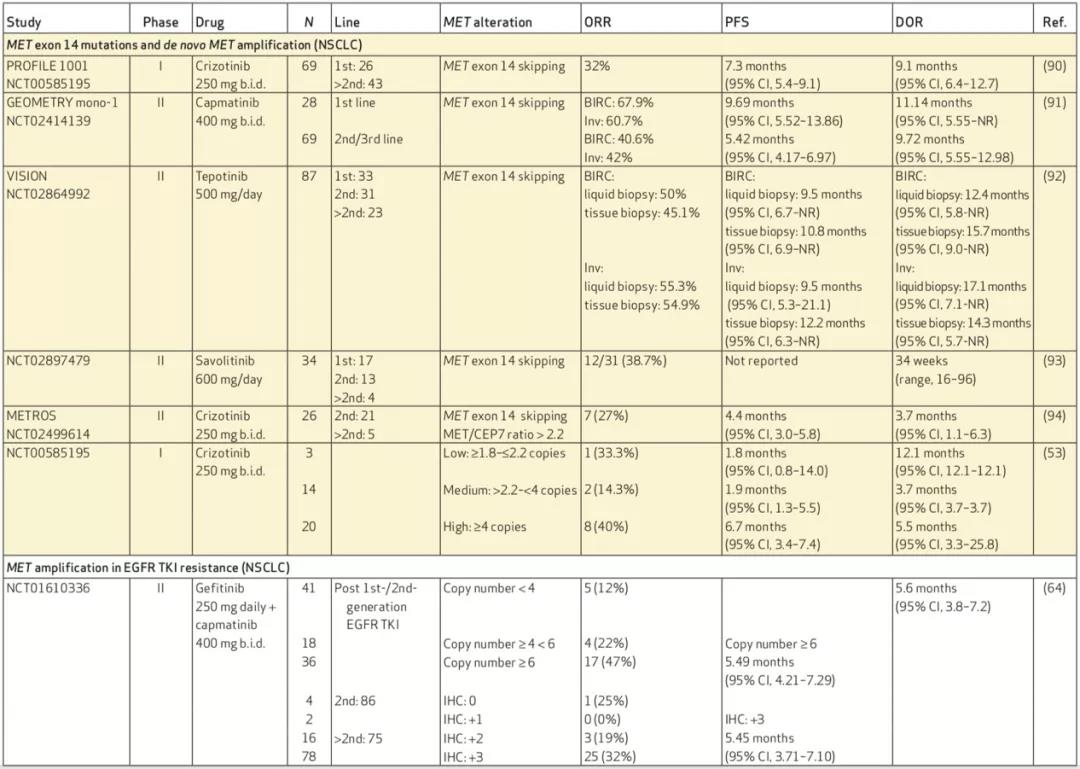
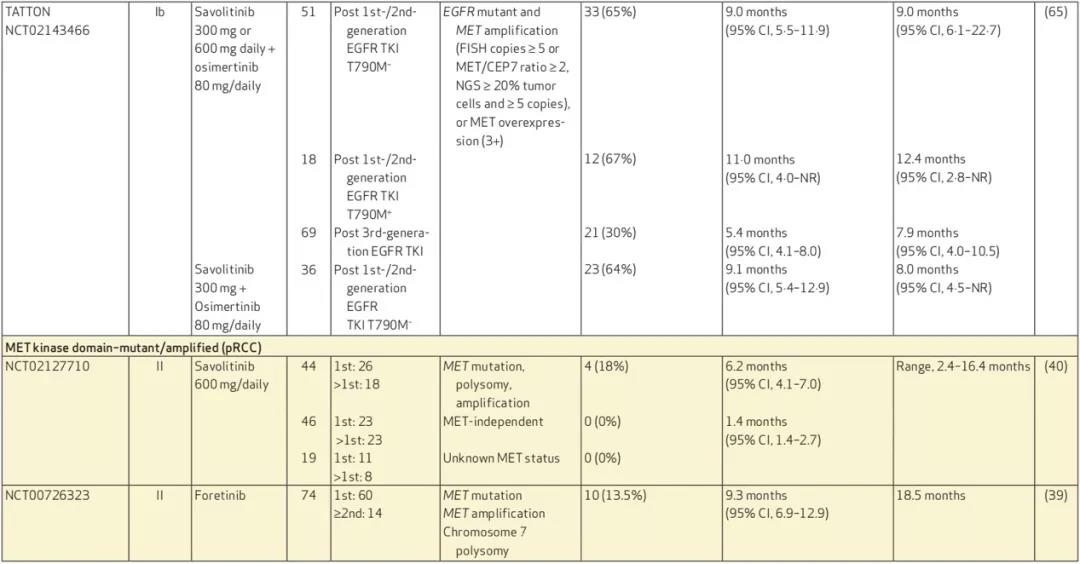
约有3%晚期NSCLC存在MET 14外显子点突变或缺失,这些变异进一步导致14外显子跳读。临床中MET 14外显子变异常见于老年患者(中位年龄72岁),有吸烟史,但在未吸烟患者中也有报道。MET 14外显子变异多见于肺腺癌,在非肉瘤样癌,鳞癌及腺鳞癌中也有发现。原发性MET 14外显子变异与其它驱动基因变异(KRAS,EGFR,ALK,ROS1和RET)相互排斥,而与TP53突变,CDKN2A/B缺失,MET,MDM2和CDK4/6扩增具有高度相关性。
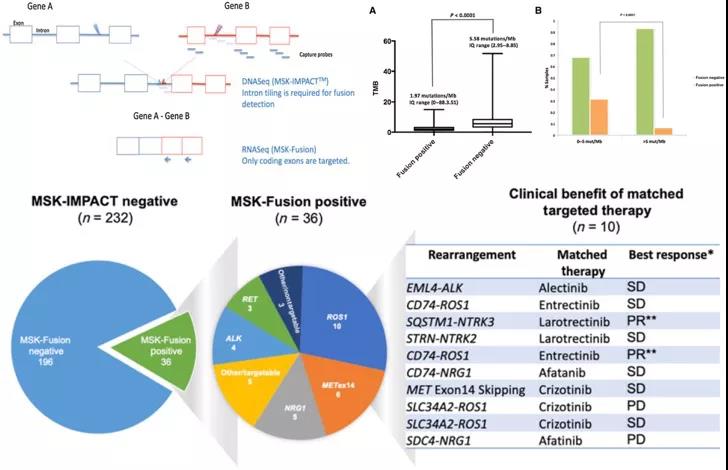
由于MET变异形式多样性,因此14外显子跳读对现有临床检测手段具有一定挑战性,目前主要是通过基于DNA二代测序技术平台,针对14外显子Sanger测序以及基于RNA技术平台如RT-PCR或RNA二代测序技术,相较而言RT-PCR和NGS比Sanger测序在14外显子跳读具有更高灵敏度。去年来自MSKCC的团队在《临床肿瘤研究》(Clinical Cancer Research)发表了一项相关研究,通过靶向RNA测序平台能有效提高DNA测序未检出驱动基因变异且肿瘤突变负荷低的肺腺癌重排变异,其中包括14外显子跳读变异(图1),进一步强调了基于RNA平台测序技术在MET 14外显子变异检测方面重要临床意义[2]。
MET 激酶区突变
MET激酶区突变介导非配体依赖性受体磷酸化及下游信号通路激活,约13%-20%I型乳头样肾细胞癌(pRCC)存在MET酪氨酸激酶区突变,具体MET激活变异包括V1092I(V1110), H1094Y/ R/L(H1112), H1124D(H1142), L1195F/V(L1213), F1200I(F1218), V1188L(V1206), Y1220I(Y1238), D1228H/N(D1246), Y1230C/D/H(Y1248), M1131T(M1149), 以及M1250T。此外由于等位基因失衡导致7号染色体多体性,pRCC具有更高MET过表达水平,提示在这类肿瘤需要其它致癌机制共同驱动自身MET通路异常活化。
MET激酶区突变本身除了可以作为部分瘤种起始驱动基因突变,同样可以介导MET-TKI治疗的耐药,包括L1195V/F,D1228X以及Y1230X突变在MET 14外显子突变肺癌中被发现介导MET-TKI耐药。在ALK融合的肿瘤中也存在相同情况,提示针对TKI耐药的突变可能会提高激酶活性。
MET扩增
野生型MET扩增定位于7号染色体,目前临床尚未有明确检测手段和/或界值去定义MET扩增,一种方法是通过原位杂交技术(FISH)评估MET拷贝数在7号染色体拷贝数占比(CEP7),这种方法可以鉴别是MET扩增还是7号染色体整体扩增导致MET多体性。针对基于CEP7界值定义,一种是把低水平扩增定义为[1.8-2.2],中度扩增(2.2-5),高度扩增定义为不低于5。目前而言MET高度扩增可以较好预测MET抑制剂疗效,并且高度提示MET通路依赖性肿瘤,因为MET低中水平扩增肿瘤同时具有更高KRAS,EGFR和ALK共突变,提示此时MET扩增并非主要致癌驱动因素。最近对于MET高度扩增CEP7界值重新调整为≥4,且同样显示出较好疗效预测作用。
为了鉴别在初治EGFR突变肺癌中MET多体性和MET扩增,还有一种方法是结合两个指标包括每个细胞MET拷贝数≥5且CEP7占比≥2,基于这一方法,在3%的病例中发现真正MET扩增,而23%的病例仅是MET多体性。
除了FISH技术以外,基于NGS测序平台也同样可以发现MET扩增,但目前针对这一技术界值定义标准仍然欠缺。
在不同瘤种中,高水平MET扩增既是疗效预测因素同样也是预后预测因素,在NSCLC中原发性MET扩增发生率约为1%,并且是早期肺癌术后预后不佳预测因素。在MET 14外显子突变的肺癌中,有15%患者同时合并MET扩增,提示肺癌中MET依赖性通路活化可以由多个不同分子事件协同驱动。
此外MET扩增也是EGFR-TKI获得性耐药比较常见机制之一,在一代EGFR-TKI中有约5%患者耐药后出现MET扩增,三代EGFR-TKI约为10%,在ALK融合NSCLC也发现MET扩增耐药机制,提示MET信号增强可以维持下游致癌信号旁路激活,进而抑制细胞凋亡和促进肿瘤增殖,目前已有多项针对MET扩增介导EGFR-TKI耐药后临床试验探索。
MET融合
MET融合在多个瘤种中均有报道,在NSCLC约为0.5%,儿童脑胶质瘤约为10%,成人脑胶质瘤为3%。目前已有报道的MET融合伴侣包括TPR, HLA-DRB1, KIF5B, PTPRZ1, STARD3NL以及ST7。既往一项个案报道发现ST7-MET融合介导一例三代ALK-TKI治疗的获得性耐药,因此对于MET融合肿瘤,MET-TKI可能是有效治疗策略。
MET过表达
MET过表达在NSCLC较为常见,经IHC确认MET过表达约占20%-25%,但目前关于MET过表达在肺癌中是否是孤立预后因素仍未盖棺定论。临床中MET过表达检测是通过SP44抗体对石蜡包埋组织进行半定量评估(0-3+),尚未明确高MET表达界值定义,部分研究将超过50%以上肿瘤细胞出现2+或3+以上定义为高水平MET过表达。基于MET过表达并未显示出能较好预测MET抑制剂疗效,提示MET过表达可能并不是有效预测生物标志物,而且MET过表达和MET扩增CEP7不具有明显相关性。在一项肺肉瘤样癌研究中基于IHC检测MET过表达在MET扩增肿瘤中灵敏度仅为50%,阳性预测率和阴性预测率分别为21.4%和94%,因此目前基于IHC检测MET过表达既不是MET异常替代指标也不是MET扩增有效筛查手段。
靶向MET变异临床治疗策略
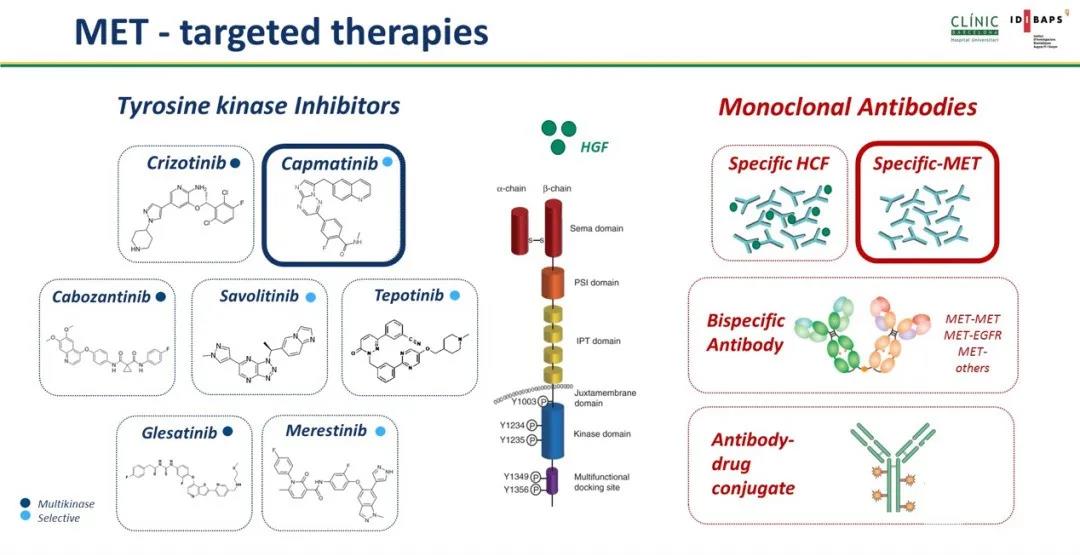
针对MET小分子激酶区抑制剂主要分为三类:I型抑制剂,主要结合ATP口袋中活性构象激酶区;II型抑制剂,主要结合在ATP口袋非活性构象激酶区;III型抑制剂,是一类非ATP竞争性变构抑制剂,主要结合于ATP口袋区以外。目前I型MET-TKI已经处于临床研发探索阶段,I型抑制剂又可细分为Ia型抑制剂包括克唑替尼以及Ib型抑制剂包括Tepotinib,Capmatinib,Savolitinib以及APL-101。II型抑制剂目前有Cabozantinib,Foretinib以及Glesatinib。除酪氨酸激酶抑制剂以外,还有其它靶向MET胞膜外区药物正在研发当中,包括复合单克隆抗体(Sym015),METxMET双特异性抗体(REGN5093),EGFR-MET双特异性抗体(JNJ-61186372)以及MET抗体偶联药物(ABBV-399, Telisotuzumab)(图3)。
目前多项临床研究正探索不同MET-TKI在MET 14外显子突变NSCLC疗效,其中I型MET抑制剂显示初步应答率从32%到68%不等,中位无进展生存期从5.4个月到12.2个月,但目前暂未有针对不同治疗线数临床研究,后续还需要进一步探索针对包括后线治疗序贯策略及其它潜在治疗策略(免疫和化疗)的临床研究。II型抑制剂包括Cabozantinib及Merestinib临床研究目前仍在进行中,关于颅内应答情况还需要更多前瞻性数据,初步研究结果显示部分MET-TKI具有一定针对颅内转移灶抗肿瘤疗效。
除MET 14外显子突变NSCLC外,MET-TKI同样也被应用于治疗MET扩增,包括原发性MET扩增和EGFR-TKI治疗后MET获得性扩增。在一项I期研究中对于MET CEP7≥4 高度扩增NSCLC接受克唑替尼治疗,40%患者达到疾病缓解,中位无进展生存为6.7个月。对于MET介导EGFR-TKI获得性耐药患者,无论是吉非替尼联合Capmatinib抑或奥西替尼联合Savolitinib均显示出较好疗效,目前SAVANNAH研究及ORCHARD研究也同样在进行EGFR-TKI耐药后MET-TKI联合治疗临床研究(图4)。
另外既往一些个案报道也发现MET-TKI(克唑替尼)对于MET融合NSCLC及原发性脑肿瘤(PLB1001/CBT-101/APL-101)具有一定活性。
而对于MET单克隆抗体临床探索则尤为曲折,在一项III期研究中,MET单抗(Onartuzumab)在非选择化疗失败后患者显示出总生存降低,既是FISH提示MET扩增患者中同样未见生存获益,此外在多个瘤种中,Onartuzumab联合化疗也未见明显预后改善,另一类似药物Emibetuzumab也在临床研发过程中因疗效不佳惨遭夭折。Rilotumumab是一类靶向可溶性HGF的单克隆抗体,在晚期胃癌及食管癌中联合化疗同样导致更差预后,这一系列早期研究缺少对患者群体选择,同时在研究中应用IHC这种尚未被证实MET检测手段,而MET单抗联合化疗导致预后更差原因推测有可能是因为抑制MET通路后异常下调免疫微环境中免疫介导的细胞毒性。
除上述药物以外,目前还有其它新型直接作用于MET靶点药物,包括像Sym015(复合IgG1人源化单克隆抗体),EGFR-MET双特异性抗体(JNJ-61186372)以及MET抗体偶联药物(ABBV-399, Telisotuzumab)等,目前这类药物仍然处于临床前或早期临床试验阶段,还有待后续进一步研究探索。
MET-TKI耐药机制
结合既往对TKI药物耐药机制探索,目前TKI耐药机制主要可以分为三类,包括激酶区获得性耐药抑制药物结合或其ATP活性,旁路信号激活以及组织学转化(小细胞肺癌转化),当然还有相当部分耐药机制仍未明。近期Pasi Janne教授等团队也汇总了MET 14外显子突变患者接受MET-TKI获得性耐药机制(图5)[3]。
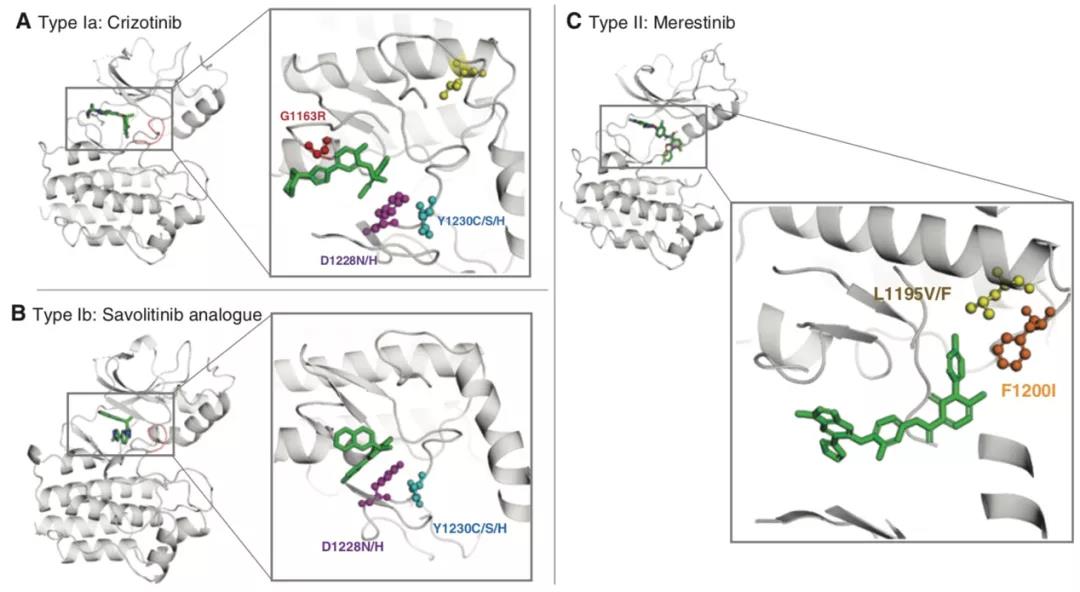
在MET扩增或MET 14外显子突变的NSCLC,继发性MET激酶区突变无论在临床前抑或临床研究中均被发现是I型及II型 MET-TKI耐药,其中D1228(D1246)和Y1230(Y1248)可以通过抑制药物和受体结合从而介导I型MET-TKI耐药,但并不影响II型MET-TKI的疗效。而G1163位点突变可以介导Ia型MET抑制剂(克唑替尼)耐药但不影响Ib型及II型MET-TKI(图6)。当使用MET-TKI进展后,往往能同时检测到多个MET突变,进一步提示针对MET-TKI耐药机制主要为单克隆源性。

