程序化阻断癌细胞EGRFR、诱导DNA损伤和抑制PARP的
今天为大家带来一篇JMC(Journalof Medicinal Chemistry ,IF=6.054)的文章,文章通讯作者为Bertrand Jean-Claude,他是麦吉尔大学健康中心(RI-MUHC)研究所CTB药物研发平台主任,代谢性疾病和并发症项目(MeDiC)副主任。麦吉尔大学的医学副教授。他的研究项目集中在一种新的肿瘤靶向方法上,这是在他的癌症药物研究实验室发起的,被称为“综合靶向概念”。这种方法寻求赋予强有力的DNA损伤剂信号抑制特性,目的是干扰导致细胞凋亡的机制。
▉ 导读
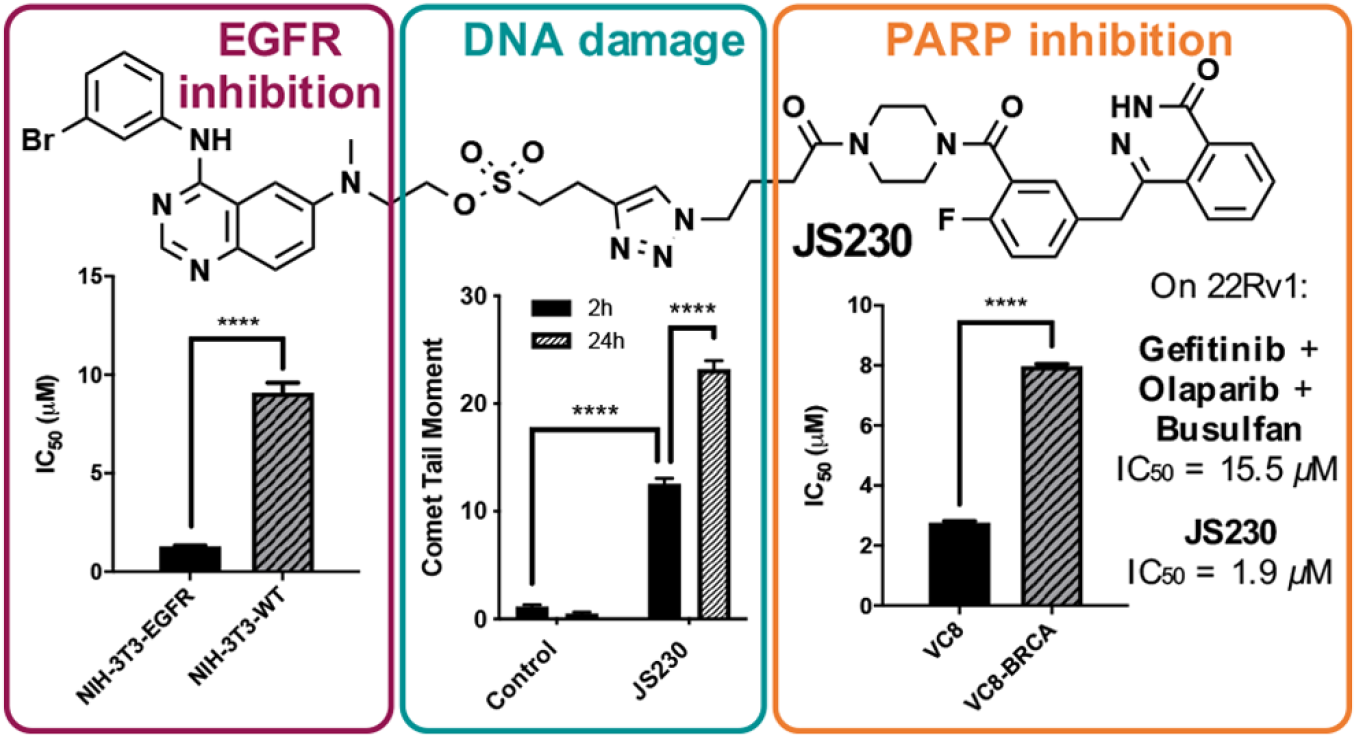
为了减少多种药物组合带来的不良反应,作者设计了一种新的可以将多种功能(如抑制EGFR磷酸化、诱导DNA损伤和阻断其修复)集中到单个分子中的组合分子JS230,它能够以剂量依赖的方式抑制EGFR(EGF receptor)、损伤DNA以及通过抑制DNA修复酶(PARP)来杀伤癌细胞。JS230的三重作用机制抑制效力优于经典的二、三联用药。
▉ 研究背景
化疗药物,根据其作用机制可以分为数种不同类型。例如,烷化剂可以直接与癌细胞的DNA反应,从而导致基因组不稳定和引发细胞死亡。然而,由于存在明显的副作用,如全身毒性、缺乏选择性和DNA修复蛋白介导的耐药性,限制了它们的临床应用。当DNA损伤时,DNA修复酶(PARP)被招募,将多个DNA修复蛋白吸引到损伤部位,从而对烷基化试剂导致的DNA损失进行修复。
PARP抑制剂(如olaparib)可导致BRCA1/2突变型肿瘤细胞的死亡,抑制PARP通常会使耐药肿瘤细胞对DNA烷化剂更敏感。有证据表明,细胞表面受体酪氨酸激酶的过度表达使DNA修复活性和抗凋亡信号增加,这会导致细胞对DNA烷化剂的耐药性,而EGFR抑制剂导致DNA碱基切除修复通路(BER)中关键分子的下调,并使细胞对烷化剂重新敏感。
针对酪氨酸受体、PARP与化疗耐药相关的不良代偿信号,作者设计了一种“组合分子”来研究其抗癌作用。

作者将这种组合分子,概括为上图中的I1−I2,其可以分成三种不同的作用机制。I:组合分子作为完整分子可以阻断靶点T1(例如EGFR),水解后则能分解为靶向T1(EGFR)的I1(EGFR抑制剂)和T2(DNA)的I2(DNA烷化剂)。II:组合分子可以分别阻断靶点T1和T2。III:机制1和2的结合形式。目前在研的分子AL776即是一种针对EGFR(T1)和Src(T2)的组合分子。
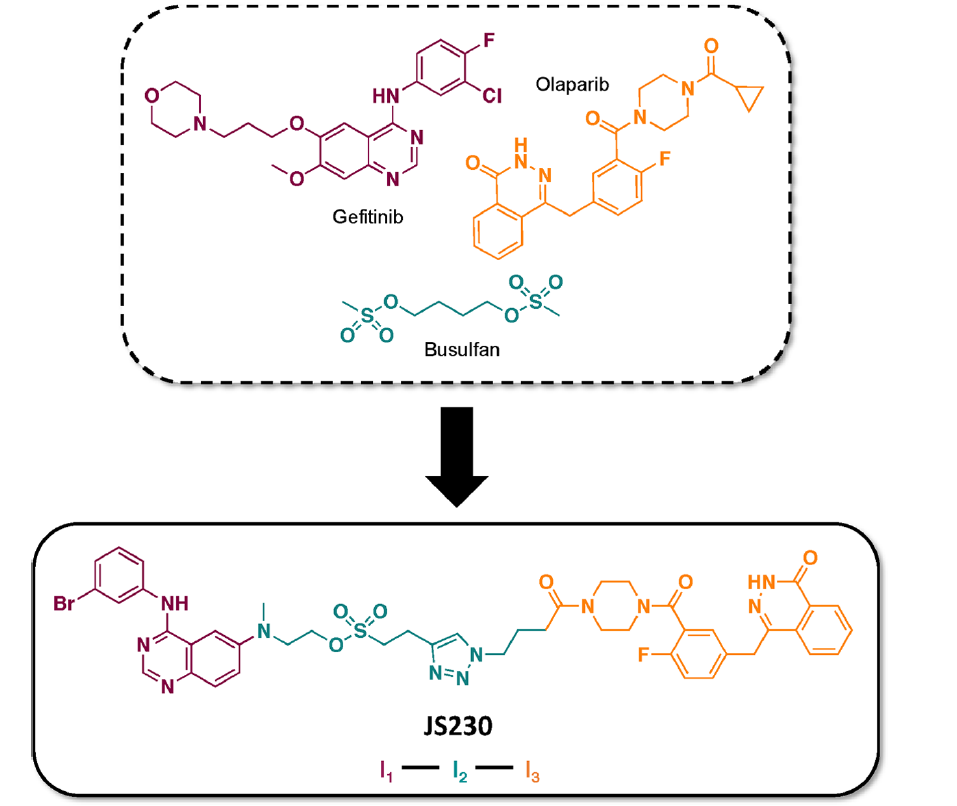
基于此策略,作者进而提出了一个三元组合分子的概念(I1−I2−I3),其包含EGFR靶向部分(I1)、DNA损伤功能部分(I2)以及DNA修复抑制(PARP)部分(I3)(阻止DNA损伤的修复)。
JS230被设计成以EGFR为靶标的喹唑啉弹头,通过烷基磺酸支链链接至一个独特的点击化学产物(如三氮唑)并延伸至PARP抑制剂olaparib上的。虽然新一代EGFR抑制剂(例如osimertinib)显示出对抗耐药EGFR突变体的独特效力,但为了限制分子的体积,避免细胞渗透性差的风险,作者选择体积较小的第一代喹唑啉抑制剂吉非替尼来建立三元分子的模型。在此,作者评估了JS230调节靶点T1(EGFR)、T2(DNA)和T3(PARP)的能力,并将其作用机制与相应的临床药物和组合进行了比较。
▉ 内容
a| 体外烷基化实验
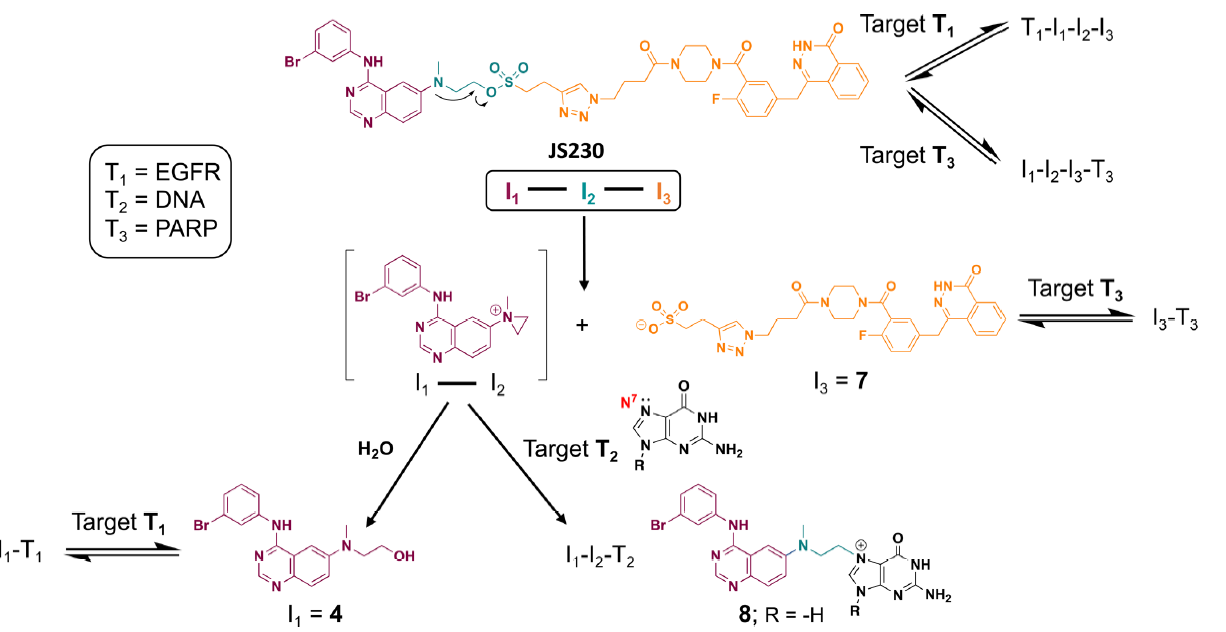
在生理条件下通过与2’脱氧鸟苷(DG)的直接化学反应来评估JS230与DNA的反应性,如图所示DNA损害剂主要与DNA中鸟嘌呤的N7位反应。同时,在反应混合物中检测到了4,该产物可能是从中间体I1−I2的水解所得。
由于在产物中发现的主要化合物4是一种EGFR抑制剂,所以可能只有一小部分JS230可能够烷化DNA。因此进一步的,作者进行了系统的靶分析以证明其在整个细胞中阻断EGFR(靶T1)、损伤DNA(靶T2)和抑制PARP(靶T3)的能力。
b| 目标1(T1):下调EGFR激活的通路
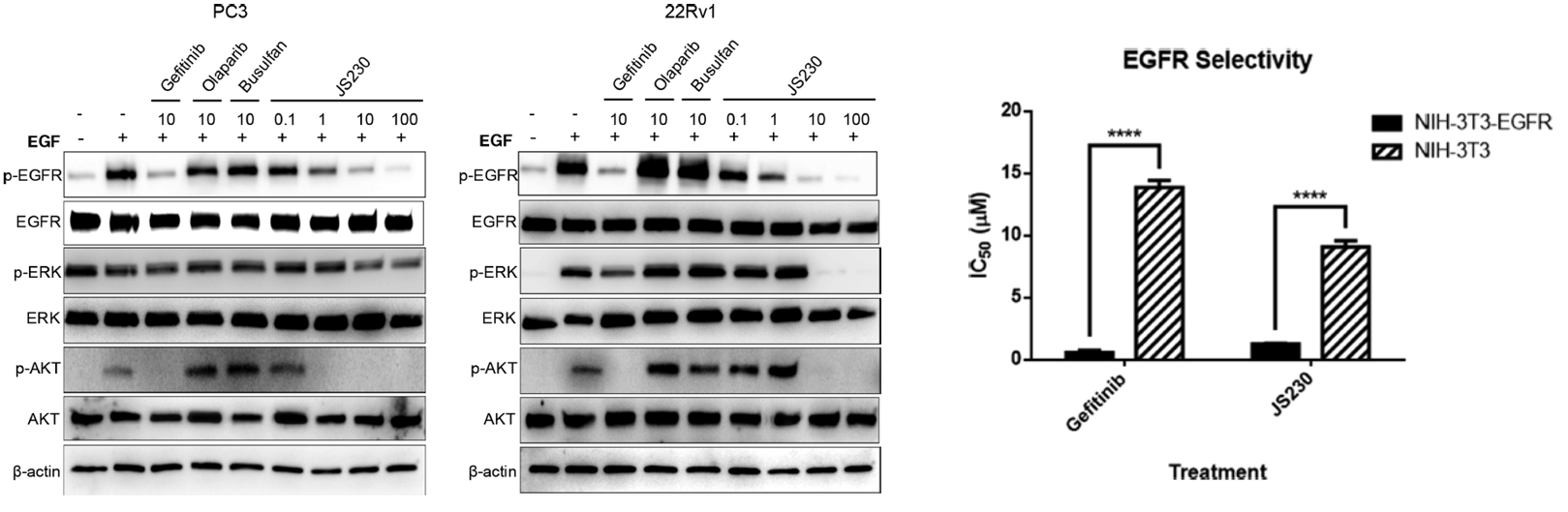
作者首先在EGF刺激的全细胞磷酸化实验中分析了JS230阻断EGFR磷酸化的能力。事实上,JS230以剂量依赖的方式抑制EGFR磷酸化,即使低至1μM浓度对PC3和22Rv1前列腺癌细胞的抑制程度为34-45%抑制作用。此外,这些细胞中ERK和AKT信号下调,表明EGFR下游的激酶功能也有所降低。最后,为了证实该分子对EGFR具有选择性,作者使用了基于细胞的选择性实验,该实验由一对同基因的细胞(转染EGFR的NIH-3T3和NIH-3T3)组成。结果表明,JS230对NIH-3T3-EGFR转染细胞具有7倍的选择性。这些实验提供了强有力的证据表明,JS230可以选择性地靶向和抑制EGFR介导的信号转导。
c| 目标2(T2):诱导DNA损伤
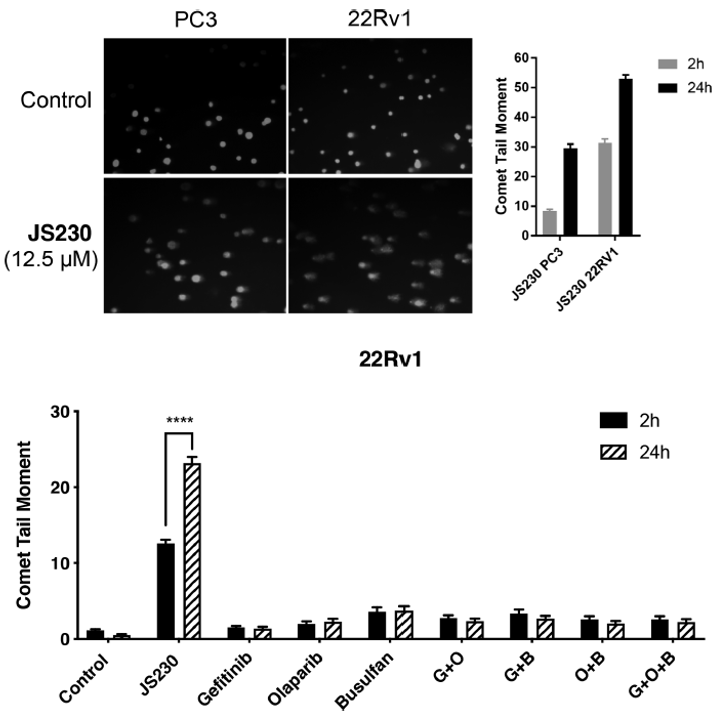
作者用碱性彗星试验评估PC3和22Rv1细胞的DNA损伤,证明了三链复合分子能够以剂量依赖的方式诱导整个细胞的DNA损伤,这与2'-脱氧鸟苷的化学反应中观察到的烷基磺酸基的烷基化能力是一致的。
d| 目标3(T3):抑制PARP
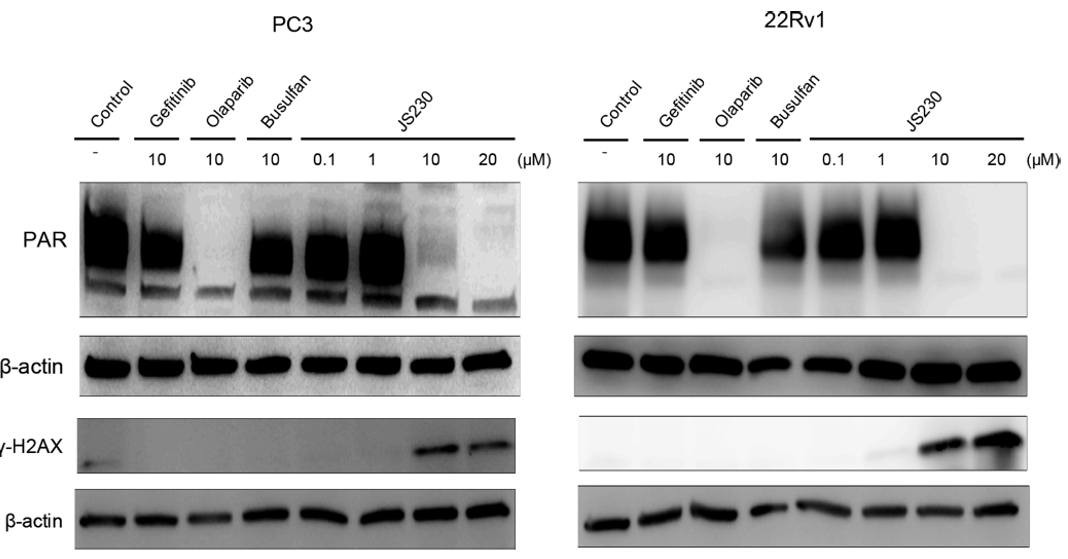
作者使用Western blot来评估JS230在整个细胞中阻断PARP的能力。结果表明,JS230对PARP有较强的抑制作用,其抑制作用与olaparib相似。
e| 生长抑制力
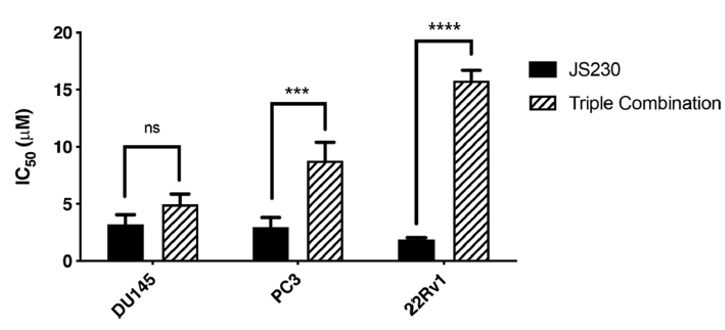
为了分析与组合分子的生长抑制效力,作者用SRB生长抑制试验比较了JS230与单一临床药物 gefitinib、olaparib、busulfan以及它们的不同等摩尔组合在EGFR过表达和olaparib耐药的前列腺癌细胞中的生长抑制效果。结果显示JS230表现出比三种单药或组合用药强2-8倍的活性,特别是在22Rv1中,JS230能够完全消除吉非替尼和olaparib单独使用或联合使用时的耐药性。
e| 细胞内定位实验
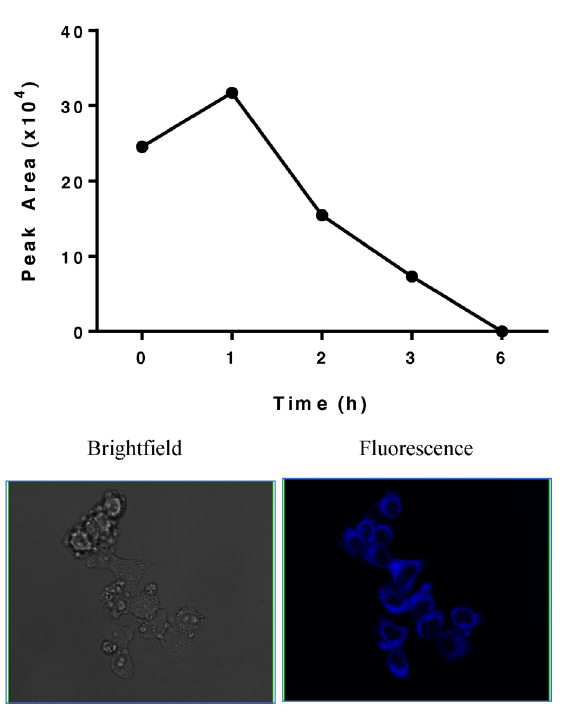
为了证实JS230确实作为一个完整的分子进入细胞的假设,对其在细胞内的水平进行了时序分析。LC−MS分析表明,JS230在1小时内达到最高细胞浓度,并至少在6小时内保持其完整的结构。另外JS230喹唑啉支架所固有的荧光特性使作者可以在整个细胞中看到组合分子,如图所示,JS230在处理后2h优先分布在核周区域,它们可能从那里扩散到细胞核,从而导致DNA损伤。因此,类似的完整的分子或其相应的活性结构必须具有核定位的能力。
▉ 总结
综上所述,本研究首次确凿地证明了设计一种三元功能分子作为双重EGFR-PARP靶向抑制剂(T1和T3)的可行性,同时造成高水平的DNA损伤(T2)。由此产生的单分子I1−I2−I3比以I1+I2+I3为代表的临床药物组合的效力显着提高(8倍),这可能是由于其独特的支架所带来的亚细胞定位和三重作用机制。这种方法可能会启发新一代分子的设计,能够在逆转化疗耐药的背景下解决肿瘤的异质性和晚期癌症的靶点多样性。
