非小细胞肺癌中MET抑制剂的耐药机制及应对策略研究进展
摘要
MET基因是非小细胞肺癌的重要肿瘤驱动基因,针对MET 14外显子跳跃突变的靶向药物为患者带来新的治疗希望。虽然以tepotinib和沃利替尼等为代表的MET抑制剂显示出良好的抗肿瘤效果,但MET抑制剂的耐药不可避免。通过对HGF/MET信号通路的研究,不仅有助于探索MET抑制剂的耐药机制,有利于找到抑制和逆转耐药的方法,而且能够扩大新药研发的领域。初步研究显示HGF/MET信号通路抑制剂与其他药物的联合应用可能具有更大的临床应用潜力。本文就MET基因异常的特点,MET抑制剂的耐药机制和应对耐药策略进行综述,并提出MET抑制剂未来的发展方向和面临的挑战。
概述
在非小细胞肺癌(non- small cell lung cancer,NSCLC)的治疗中,针对肿瘤驱动基因的靶向药物层出不穷。如口服小分子酪氨酸激酶抑制剂(tyrosine kinase inhibitors,TKIs)已被批准用于表皮生长因子受体(epidermal growth factor receptor,EGFR)突变、间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)重排和ROS1重排的NSCLC[1-2]。间质-上皮细胞转化因子(mesenchymal-epithelial transition factor,MET)被认为是继EGFR、ALK和ROS1之后另一个重要的肿瘤驱动基因,针对MET基因突变的靶向药物受到越来越多的关注。与MET基因相关的异常状态主要包括MET 14外显子跳跃突变、MET基因扩增和蛋白过表达。其中,针对MET 14外显子跳跃突变的靶向药物发展最快,已经上市和即将上市的药物包括克唑替尼、cabozantinib、沃利替尼、tepotinib、capmatinib等,另外还有许多药物正在进行临床研究[3-4]。但是,与其他靶向药物一样,MET抑制剂的耐药不可避免,给患者的治疗带来巨大的挑战。本文将结合MET基因异常的特点,重点对MET抑制剂的耐药机制和应对策略进行综述,并提出未来MET抑制剂的发展方向和面临的挑战。
01
MET基因突变
1.1 MET基因和HGF/MET信号通路
MET基因位于人类7号染色体(7q21-31),长度约125 kb,同时含有21个外显子[5]。由MET基因编码的蛋白为c-MET,也称为肝细胞生长因子受体(hepatocyte growth factor receptor,HGFR),是具有自主磷酸化活性的跨膜受体,属于酪氨酸激酶受体超家族,主要表达于上皮细胞。肝细胞生长因子(hepatocyte growth factor,HGF)是目前发现的唯一的c-MET配体,属于纤维蛋白溶酶原家族,主要表达于间质细胞。HGF能够与c-MET的细胞外结构域结合,促使c-MET发生二聚化、酪氨酸磷酸化,激活众多下游信号通路,如PI3K-Akt、RasMAPK、STAT和Wnt/β-catenin等,从而发挥促进细胞增殖、细胞生长、细胞迁移、侵袭血管及血管生成等效应。c-MET的结构和功能见图 1。c-MET正常表达时促进组织的分化与修复,当存在异常时则可能促进肿瘤的增殖与转移[6]。HGF/MET信号通路异常激活主要包括MET 14外显子跳跃突变、MET基因扩增和c-MET蛋白过表达。
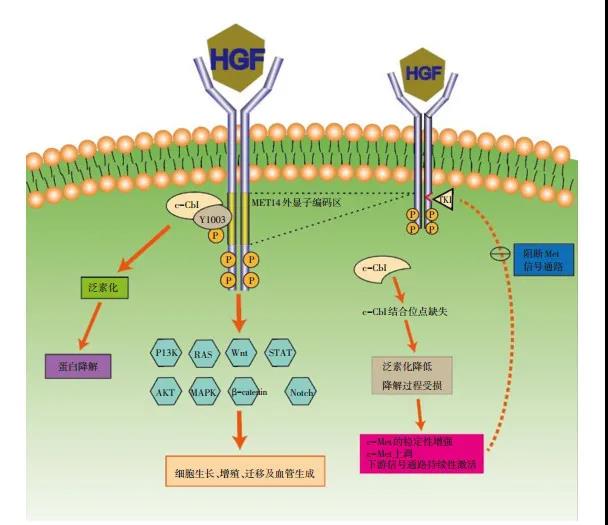
1.2 MET 14外显子跳跃突变
c-MET主要由E3泛素连接酶c-Cbl主导降解。MET 14外显子对应编码141个氨基酸,其所在的近膜结构域是c-MET的关键负性调控区,包含E3泛素连接酶c-Cbl酪氨酸结合位点(Y1003),参与c-MET蛋白的泛素化和降解。MET 14外显子的基因突变会引起14外显子跳读(exon skipping),使得含有E3泛素连接酶c-Cbl结合位点的近膜结构域缺失,进而导致c-MET蛋白泛素化障碍、c-MET稳定性增加和降解率降低,引起下游信号的持续激活,最终成为肿瘤的驱动基因。MET 14外显子跳跃突变的机制见图 1。在NSCLC中,MET 14外显子跳跃突变的总体发生率为3%~6%[7-8],并且不与EGFR、ALK等NSCLC的其他驱动基因共存,提示其代表一种孤立的肿瘤驱动基因[9-10]。但MET 14外显子跳跃突变可以与MET基因扩增和蛋白过表达并存[11]。
1.3 MET基因扩增
MET基因扩增即MET基因的拷贝数增加,包括整体染色体重复和局部区域基因重复[12],其中整体染色体重复是指肿瘤细胞中出现多条7号染色体。MET基因扩增通常伴有EGFR、KRAS等其他基因突变,有研究显示MET扩增可能并不是NSCLC的肿瘤驱动基因[13]。MET扩增与EGFR、KRAS等其他驱动基因的激活有明确的联系,可能是EGFR基因突变的NSCLC获得性耐药的机制之一。有研究显示,15%~20%的EGFR获得性耐药患者可检测到MET扩增[14-15]。另外,MET基因扩增往往提示NSCLC患者的预后较差[11]。
1.4 c-MET过表达或HGF过表达
HGF与c-MET结合可引起下游信号通路的激活,因此若存在c-MET或HGF过表达的异常情况,可能导致下游信号通路的持续激活,进而导致肿瘤的发生和发展。有研究表明,NSCLC中HGF/c-Met过表达与淋巴管生成密切相关[16]。c-MET过表达在肺腺癌中的发生率可高达65%,但其中仅10%的c-MET过表达伴有MET基因突变,因此c-MET过表达可能并不是原发致癌驱动因素,更可能是作为其他驱动基因激活后产生的二次事件,从而促进肿瘤的生长[17]。
02
MET抑制剂的作用机制
基于HGF/c-MET信号通路异常激活,MET抑制剂成为NSCLC的重要治疗手段。跟据HGF/c-MET信号通路中作用位点的不同,可将MET抑制剂分为3大类:抗HGF单克隆抗体、抗c-MET单克隆抗体和小分子TKI。前两者分别在细胞外与HGF和c-MET结合,从而阻止HGF与c-MET的结合及受体磷酸化,阻止信号传导;小分子MET-TKI作用于膜内催化域从而阻止蛋白磷酸化,阻断信号传导(图 1)。目前研究最多且最具有治疗潜力的是小分子MET-TKI。
2.1 MET-TKI
MET-TKI可分为3种类型(Ⅰ型、Ⅱ型和Ⅲ型)[18]。Ⅰ型TKI是ATP竞争性抑制剂,与MET主链中的氨基酸残基形成氢键,其中又分为Ⅰa型和Ⅰb型,Ⅰb型TKI可以结合的位点较少,不包括甘氨酸残基的G1163位点(类似于ALK的G1202和ROS1的G2032位点),因此特异性较高。临床常用的药物克唑替尼属于Ⅰa型METTKI,tepotinib、沃利替尼和AMG337等均属于Ⅰb型METTKI。Ⅱ型MET-TKI一般为多靶点TKI,不仅作用于ATP结合位点,还能通过管家基因突变进入非活性DFG-out构象形成的疏水口袋,对产生二次突变的MET仍具有抑制作用,或许可以逆转由Y1230等突变引起的Ⅰ型MET-TKI耐药[3]。cabozantinib属于Ⅱ型MET-TKI。Ⅲ型MET-TKI作用于与ATP结合位点完全不同的变构位点,目前尚无药物进入临床研究阶段。
2.2 抗HGF/c-MET单克隆抗体
该类药物的原理是通过单克隆抗体竞争性结合HGF或c-MET,阻断HGF和c-MET之间的相互结合。此类药物包括rilotumumab(AMG-102)、ficlatuzumab(AV-299)和TAK-701等[19-21]。抗c-MET单克隆抗体与c-MET结合,一方面阻断了HGF与c-MET的结合,另一方面此类受体与配体在细胞表面的结合不引发c-MET信号传导,无法诱导c-MET二聚化。遗憾的是,部分抗HGF/c-MET单克隆抗体的临床试验失败,部分仍在进行中,目前尚无成功的抗HGF/c-MET单克隆抗体药物上市。
03
MET抑制剂的耐药机制
MET抑制剂的耐药机制研究主要集中在METTKI药物,可分为原发性耐药和继发性耐药。
3.1 原发性耐药
MET-TKI通过特定氨基酸上的疏水相互作用而与c-MET的ATP口袋紧密结合。在MET作为致癌驱动因素的肿瘤中,用MET-TKI进行的体外诱变分析已鉴定出针对Ⅰ型MET抑制剂的几种优势耐药突变(Y1230,D1228)和部分轻微耐药突变(F1200,V1155)[22-23]。
Fujino等[24]对8种MET抑制剂进行的体外试验研究显示,MET基因中D1288和Y1230突变可能导致Ⅰ型MET-TKI耐药,L1195和F1200突变可能导致Ⅱ型MET-TKI耐药,但Ⅰ型和Ⅱ型MET-TKI之间无交叉耐药现象。另外,有研究显示HGF/MET的下游信号通路改变,如PI3K信号通路的改变,是MET抑制剂原发耐药的机制之一[25]。
3.2 继发性耐药机制
在应用MET抑制剂初始治疗有效后,可能出现新的MET结构域改变,从而导致继发性耐药的发生。Dong等[26]报道1例MET 14外显子突变的NSCLC患者在应用克唑替尼治疗后耐药,二代测序显示外周血循环肿瘤细胞同时出现MET基因D1228N/H和Y1230H突变。另有研究报道MET 14外显子突变的NSCLC患者接受克唑替尼治疗后,产生获得性耐药突变MET D1228N和MET Y1230C[27-28]。Bahcall等[29]研究显示MET基因中D1228V点突变可能是沃利替尼继发性耐药的原因之一。韩森等[30]报道了1例MET 14外显子突变的晚期肺肉瘤样癌患者应用沃利替尼治疗后出现耐药,二次活检提示新出现了成纤维细胞生长因子受体(fibroblast growth factor receptor 1,FGFR1)、EGFR和KRAS的基因扩增。Li等[31]研究发现MET基因中Y1248H和D1246N突变是MET抑制剂继发性耐药的原因之一。另外,GimenezXavier等[32]从基因组学的角度进行研究发现,2型神经纤维瘤病(neurofibromatosis type 2,NF2)基因对于MET抑制剂的继发性耐药起到关键作用。
总之,MET抑制剂的耐药机制复发多样,既包括了原有MET基因原发性改变,也包括在MET-TKI用药后出现的新突变;既包括其他相关基因的扩增或激活,也包括HGF/MET下游信号通路的改变等(图 2)。
04
抑制或逆转MET抑制剂耐药的方法
4.1 不同类型的MET-TKI之间的互换
Ⅰ型MET-TKI需要与Y1230堆叠才能结合cMET,Y1230突变降低了Ⅰ型MET- TKI的结合能力。从Ⅰ型MET-TKI转换为Ⅱ型有可能克服此类耐药突变。如1例MET阳性晚期肺腺癌患者在应用沃利替尼治疗后进展,发现新的D1228V点突变,后改为cabozantinib治疗有效[29]。不同类型的MET-TKI结合位点有所不同,所以耐药机制并不完全相同,因此存在一定的非交叉耐药特点,恰当的二次活检和基因检测可能有助于耐药后选择更为合适的药物。目前认为Ⅰ型MET-TKI和Ⅱ型MET-TKI在用药顺序上无明显的优劣,若Ⅰ型MET-TKI用药后发生耐药,则可以考虑换为Ⅱ型MET-TKI,若Ⅱ型MET-TKI发生耐药,则可以考虑换为Ⅰ型,均可能再次产生疗效。抑制或逆转耐药的机制见图 2。
4.2 HGF/MET信号通路的多重阻断
在HGF/MET信号通路中,联合应用抗HGF/c-MET单克隆抗体和MET-TKI可能更有效地阻断下游信号通路的激活,从而达到逆转耐药的效果。虽然这一假设在理论上有效,但需要进一步的临床试验验证。
4.3 与免疫治疗的结合
近年来免疫治疗在NSCLC领域取得了突飞猛进的发展。Rivzi等[33]最先报道肿瘤突变负荷(tumor muta⁃ tional burden,TMB)与NSCLC患者对帕博丽珠单抗的治疗反应密切相关。MET 14外显子突变的NSCLC患者的平均TMB为6.9突变/Mb(范围0~197.9),低于肺癌总体人群(平均值为10.7突变/Mb),但高于EGFR突变患者(平均值为4.5突变/Mb)和ALK阳性患者(平均值为2.8突变/Mb)[34-35]。EGFR突变和ALK阳性的NSCLC患者,PD-L1表达较低,对抗PD-L1/PD-1药物的治疗效果也较差[36]。虽然MET基因突变患者的PD-L1表达情况尚不明确,但是考虑到TMB的情况,MET抑制剂与免疫治疗的联合有可能克服耐药。国内的一项研究显示,c-MET/PD-1双抗在体外试验中能够有效抑制c-MET和PD-L1均高表达的肺癌细胞的生长、迁移和抗凋亡作用[37]。另外有研究显示,MET抑制剂能够上调肺腺癌PD-L1的表达,HFGF/MET信号通路和免疫逃逸存在一定的关联性[38-39],这也为MET抑制剂和免疫治疗的联合应用提供了理论依据。
4.4 HGF/MET下游信号通路抑制剂
最新的研究发现,PI3K信号通路的改变可能是MET抑制剂原发性耐药的原因。Jamme等[25]对65例MET 14外显子突变的晚期NSCLC患者进行研究,发现其中2例患者存在PIK3CA突变,6例患者存在PTEN缺失。存在PI3K信号通路改变的3例患者在接受MET-TKI治疗后均提示肿瘤进展。另外,METTKI对存在PI3K途径改变的MET 14外显子突变细胞系(包括PTEN缺失患者来源的细胞系)的增殖无抑制作用。该研究同时提示MET-TKI与PI3K抑制剂联合治疗可抑制PI3K和MAPK信号转导,并恢复对MET-TKI的敏感性。因此,为克服MET-TKI耐药提供了新的治疗思路。
05
结语
MET基因是NSCLC的重要肿瘤驱动基因,METTKI是治疗NSCLC患者中具有MET基因突变人群的有效药物。但是MET-TKI的耐药不可避免,HGF/ MET信号通路的相关研究,对于抑制和逆转METTKI的耐药具有重要意义。MET-TKI与其他药物的联合应用,可能是未来研究的方向。总之,在肺癌的精准治疗时代,分子检测和靶向药物必将为存在HGF/MET信号通路异常的NSCLC患者带来更多的治疗机会和更好的疗效。
End
参考文献:
[1]
Joshua K, Fernando S, Isabella B, et al. Changing the therapeutic landscape in non- small cell lung cancers:the evolution of comprehensive molecular profiling improves access to therapy[J]. Curr Oncol Rep, 2017, 19(4): 24. DOI:10.1007/s11912-017-0587-4
[2]
Anne S, Giorgio V, Paul A, et al. Scientific advances in lung cancer 2015[J]. J Thorac Oncol, 2016, 11(5): 613-638. DOI:10.1016/j.jtho.2016.03.012
[3]
Reungwetwattana T, Liang Y, Zhu V, et al. The race to target MET exon 14 skipping alterations in non-small cell lung cancer:The why, the how, the who, the unknown, and the inevitable[J]. Lung Cancer, 2017, 103: 27-37. DOI:10.1016/j.lungcan.2016.11.011
[4]
Nele V, Elisa G, Patrick P, et al. cMET Exon 14 Skipping:From the Structure to the Clinic[J]. J Thorac Oncol, 2016, 11(9): 1423-1432. DOI:10.1016/j.jtho.2016.05.005
[5]
Organ S, Tsao M. An overview of the c-MET signaling pathway[J]. Ther Adv Med Oncol, 2011, 3(Suppl 1): S7-S19.
[6]
Moosavi F, Giovannetti E, Saso L, et al. HGF/MET pathway aberrations as diagnostic, prognostic, and predictive biomarkers in human cancers[J]. Crit Rev Clin Lab Sci, 2019, 56(8): 533-566. DOI:10.1080/10408363.2019.1653821
[7]
Gow C, Hsieh M, Wu S, et al. A comprehensive analysis of clinical outcomes in lung cancer patients harboring a MET exon 14 skipping mutation compared to other driver mutations in an East Asian population[J]. Lung Cancer, 2017, 103: 82-89. DOI:10.1016/j.lungcan.2016.12.001
[8]
Heist R, Shim H, Gingipally S, et al. MET exon 14 skipping in non-small cell lung cancer[J]. Oncologist, 2016, 21(4): 481-486. DOI:10.1634/theoncologist.2015-0510
[9]
Cortot AB, Kherrouche Z, Descarpentries C, et al. Exon 14 deleted MET receptor as a new biomarker and target in cancers[J]. J Natl Cancer Inst, 2017, 109(5): 262.
[10]
Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors[J]. Cancer Discov, 2015, 5(8): 850-859. DOI:10.1158/2159-8290.CD-15-0285
[11]
Tong JH, Yeung SF, Chan AWH, et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis[J]. Clin Cancer Res, 2016, 22(12): 3048-3056. DOI:10.1158/1078-0432.CCR-15-2061
[12]
Kawakami H, Okamoto I, Okamoto W, et al. Targeting MET amplification as a new oncogenic driver[J]. Cancers(Basel), 2014, 6(3): 1540-1552. DOI:10.3390/cancers6031540
[13]
Schildhaus HU, Schultheis AM, Rüschoff J, et al. MET amplification status in therapy-naive adeno- and squamous cell carcinomas of the lung[J]. Clin Cancer Res, 2015, 21(4): 907-915. DOI:10.1158/1078-0432.CCR-14-0450
[14]
Drilon A, Cappuzzo F, Ou SH, et al. Targeting MET in lung cancer:will expectations finally be MET?[J]. J Thorac Oncol, 2017, 12(1): 15-26. DOI:10.1016/j.jtho.2016.10.014
[15]
Engelman J, Zejnullahu K, Mitsudomi K, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling[J]. Science, 2007, 316(5827): 1039-1043. DOI:10.1126/science.1141478
[16]
Zhang N, Xie F, Gao W, et al. Expression of hepatocyte growth factor and c- Met in non- small- cell lung cancer and association with lymphangiogenesis[J]. Mol Med Rep, 2015, 11(4): 2797-2804. DOI:10.3892/mmr.2014.3071
[17]
Yeung SF, Tong JHM, Law PPW, et al. Profiling of oncogenic driver events in lung adenocarcinoma revealed MET mutation as independent prognostic factor[J]. J Thorac Oncol, 2015, 10(9): 1292-1300. DOI:10.1097/JTO.0000000000000620
[18]
Gherardi E, Birchmeier W, Birchmeier C, et al. Targeting MET in cancer:rationale and progress[J]. Nat Rev Cancer, 2012, 12(2): 89-103. DOI:10.1038/nrc3205
[19]
Tarhini AA, Rafique I, Floros T, et al. Phase 1/2 study of rilotumumab(AMG 102), a hepatocyte growth factor inhibitor, and erlotinib in patients with advanced non-small cell lung cancer[J]. Cancer, 2017, 123(15): 2936-2944. DOI:10.1002/cncr.30717
[20]
Mok TSK, Sarayut LG, SU WC, et al. A randomized phase 2 study comparing the combination of ficlatuzumab and gefitinib with gefitinib alone in asian patients with advanced stage pulmonary adenocarcinoma[J]. J Thorac Oncol, 2016, 11(10): 1736-1744. DOI:10.1016/j.jtho.2016.05.038
[21]
Okamoto W, Okamoto I, Tanaka K, et al. TAK- 701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutation[J]. Mol Cancer Ther, 2010, 9(10): 2785-2792. DOI:10.1158/1535-7163.MCT-10-0481
[22]
Qi J, McTigue MA, Rogers A, et al. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors[J]. Cancer Res, 2011, 71(3): 1081-1091. DOI:10.1158/0008-5472.CAN-10-1623
[23]
Tiedt R, Degenkolbe E, Furet P, et al. A drug resistance screen using a selective MET inhibitor reveals a spectrum of mutations that partially overlap with activating mutations found in cancer patients[J]. Cancer Res, 2011, 71(15): 5255-5264. DOI:10.1158/0008-5472.CAN-10-4433
[24]
Fujino T, Kobayashi Y, Suda K, et al. Sensitivity and resistance of MET exon 14 mutations in lung cancer to eight MET tyrosine kinase inhibitors in vitro[J]. J Thorac Oncol, 2019, 14(10): 1753-1765. DOI:10.1016/j.jtho.2019.06.023
[25]
Jamme P, Fernandes M, Copin MC, et al. Alterations in the PI3K pathway drive resistance to MET inhibitors in NSCLC harboring MET exon 14 skipping mutations[J]. J Thorac Oncol, 2020, 15(5): 741-751. DOI:10.1016/j.jtho.2020.01.027
[26]
Dong HJ, Li P, Wu CL, et al. Response and acquired resistance to crizotinib in Chinese patients with lung adenocarcinomas harboring MET Exon 14 splicing alternations[J]. Lung Cancer, 2016, 102: 118-121. DOI:10.1016/j.lungcan.2016.11.006
[27]
Heist RS, Sequist LV, Borger D, et al. Acquired resistance to crizotinib in NSCLC with MET exon 14 skipping[J]. J Thorac Oncol, 2016, 11(8): 1242-1245. DOI:10.1016/j.jtho.2016.06.013
[28]
Ou SI, Young L, Schrock AB, et al. Emergence of preexisting MET Y1230C mutation as a resistance mechanism to crizotinib in NSCLC with MET exon 14 skipping[J]. J Thorac Oncol, 2017, 12(1): 137-140. DOI:10.1016/j.jtho.2016.09.119
[29]
Bahcall M, Sim T, Paweletz CP, et al. Acquired METD1228V mutation and resistance to MET inhibition in lung cancer[J]. Cancer Discov, 2016, 6(12): 1334-1341. DOI:10.1158/2159-8290.CD-16-0686
[30]
Han S, Fang J, Lu S, et al. Response and acquired resistance to savolitinib in a patient with pulmonary sarcomatoid carcinoma harboring MET exon 14 skipping mutation:a case report[J]. Onco Targets Ther, 2019, 12: 7323-7328. DOI:10.2147/OTT.S210365
[31]
Li A, Yang J, Zhang XC, et al. Acquired MET Y1248H and D1246N mutations mediate resistance to MET inhibitors in non-small cell lung cancer[J]. Clin Cancer Res, 2017, 23(16): 4929-4937. DOI:10.1158/1078-0432.CCR-16-3273
[32]
Gimenez-Xavier P, Pros E, Bonastre E, et al. Genomic and molecular screenings identify different mechanisms for acquired resistance to MET inhibitors in lung cancer cells[J]. Mol Cancer Ther, 2017, 16(7): 1366-1376. DOI:10.1158/1535-7163.MCT-17-0104
[33]
Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer[J]. Science, 2015, 348(6230): 124-128. DOI:10.1126/science.aaa1348
[34]
Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations[J]. J Thorac Oncol, 2016, 11(9): 1493-1502. DOI:10.1016/j.jtho.2016.06.004
[35]
Spigel DR, Schrock AB, Fabrizio D, et al. Total mutation burden(TMB) in lung cancer(LC) and relationship with response to PD-1/PD- L1 targeted therapies[J]. J Clin Oncol, 2016, 34(suppl 15): 9017.
[36]
Gainor JF, Shaw AT, Sequist LV, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer:a retrospective analysis[J]. Clin Cancer Res, 2016, 22(18): 4585-4593. DOI:10.1158/1078-0432.CCR-15-3101
[37]
Sun ZJ, Wu Y, Hou WH, et al. A novel bispecific c-MET/PD-1 antibody with therapeutic potential in solid cancer[J]. Oncotarget, 2017, 8(17): 29067-29079. DOI:10.18632/oncotarget.16173
[38]
Sun X, Li CW, Wang WJ, et al. Inhibition of c-MET upregulates PD-L1 expression in lung adenocarcinoma[J]. Am J Cancer Res, 2020, 10(2): 564-571.
[39]
Titmarsh HF, O'Connor R, Dhaliwal K, et al. The emerging role of the cMET-HGF axis in non-small lung cancer tumor immunology and immunotherapy[J]. Front Oncol, 2020, 10: 54. DOI:10.3389/fonc.2020.00054
