天使人综合征
天使人综合症指的是患者体内细胞的15号染色体包含的某些基因丧失遗传表达功能。缺失基因的区域位于该染色体长臂,在医学上被指定为:15q11-q13。此区域还包含一个名为UBE3A的基因,如果其变异或是缺失,很可能会引起天使人综合症患者典型的脑神经异常症状。正常人从父源和母源各遗传到UBE3A基因的一份拷贝,而此基因的这两份拷贝在机体的许多组织中,都可被正常激活。然而,在脑的特定区域,仅母源基因拷贝具有活性。此与父源或母源有关的基因激活过程与“基因组印记”有关。如果由于染色体或是基因突变而使得母源拷贝缺失,那么该患者在脑部的某些部位可能会缺失原本应发挥正常功能的UBE3A基因拷贝。在大多数情况下(约70%的病例),天使人综合症是由15号染色体的母源拷贝的遗传物质缺失而诱发的,主要是缺失UBE3A基因。因为,UBE3A基因的一份父源拷贝,通常情况下,在患者脑部的某特定区域并不具有活性,而15号染色体母源拷贝上的遗传物质缺失,主要指的是缺失 UBE3A基因的活性拷贝。在天使人综合征的3%到7%的病例中,当该患者遗传到15号染色体的2份父源拷贝,而非母源父源拷贝各一份时,就会患上天使人综合症。此现象被称为父源单亲二体症(UPD)。此病患者体内的15号染色体有两份UBE3A基因拷贝,但是它们都属于父源,因而在脑部不具有活性。约10%的天使人综合征是由UBE3A基因突变而诱发的,而另外3%是由于DNA中控制UBE3A基因和15号染色体母源拷贝上的其他基因活性的遗传物质突变而诱发的。在较少的病例中,天使人综合征是由染色体重组易位或是由除UBE3A以外的其他基因的变异而诱发的。这些基因的突变会使得 UBE3A基因非正常地失去活性。

发病率
据科学统计,此病的发病率是1.2到2万分之1。
基因突变
天使人综合征患者的某些特定症状是由于一种被称为UBE3A的基因功能发生缺失而导致的。正常人体内有父源母源UBE3A基因拷贝各一份。在许多人的机体组织中,此基因的两份拷贝都具有活性。然而,在大脑的特定区域,仅仅此基因的母源拷贝保有活性。此类与父源或母源有关的基因激活过程是由一种被称为基因组印记的现象而引发的。如果因为染色体变异或是基因突变而使得UBE3A基因的母源拷贝发生缺失,那么此患者在大脑的特定部位并没有此基因具有活性的拷贝。一些独特的遗传机理会使得UBE3A基因的母源拷贝失去活性或发生缺失。大多数天使人综合征的病例(约70%)是由于患者体内母源15号染色体包含此基因的部分片段发生缺失而诱发的。对于其他一些病例而言(约占所有病例的11%),天使人综合征是由UBE3A基因的母源拷贝变异而诱发的。在少数病例中,天使人综合症是由于患者有两份父源15号染色体拷贝而引发的,而非正常情况下,机体有一份父源,一份母源15号染色体拷贝。此现象在医学上被称为父源单亲二倍体。在相对较少的情况下,染色体重组易位以及控制UBE3A基因激活过程的DNA区域中的遗传物质发生缺失或是变异,也可能诱发天使人综合征。这些遗传物质的变异能使得15号染色体母源拷贝上的UBE3A或是其他基因失去活性。对于10%--15%的天使人综合症患者而言,仍不清楚他们的发病病因,可能是其他基因或是染色体突变导致的。对于某些天使人综合症患者而言,缺失OCA2基因可能会使其拥有浅色头发和过白的皮肤。部分此遗传病患者体内的15号染色体的某个片段上往往缺失OCA2基因。然而,缺失OCA2基因并不必然使得天使人综合征患者出现其他一些典型症状。此基因合成的蛋白基本上对皮肤,头发和眼睛瞳孔的颜色起着决定性作用。
遗传模式
大多数的天使人综合症病例并不具有遗传性,由于父源单亲二倍体现象兼有母源15号染色体遗传物质缺失而诱发的天使人综合症更是如此。在患者生殖细胞的形成过程中,或是在其胚胎的早期发育过程中,这些遗传物质的变异都是作为随机事件而发生的。通常情况下,患者的家属并没有此病的患病史。较少情况下,某类诱发天使人综合征的遗传物质突变现象具有遗传性质。例如,UBE3A基因或是其附近区域控制基因激活过程的DNA遗传物质的突变现象即可能具有遗传性。
什么是angelman综合症
angelman综合症(AS)也叫天使人综合征(症)或快乐木偶(综合)症,是一种先天性的常染色体显性遗传疾病,这种疾病影响神经系统的发育,会导致严重的智力缺陷和学习障碍。不过通常来说,AS患者的寿命和正常人相近,但他们一生都需要特殊照顾。
1965年,英国内科医生哈里·安格曼(Harry Angelman)在医学文献中首次描述了该病的症状,因此该病也被命名为Angelman综合征(症)。通常,这类患者在出生时临床特征并不明显,多数患者是在2-5岁时,由于出现特征明显的临床症状(如愉悦的面部表情和显著的智力障碍)而确诊的。据估计,每2万名儿童中就有一名患有angelman综合症。
angelman综合症的病因,现在已经明确为15号染色体上的UBE3A基因缺陷所致,其临床典型特征为愉悦的面部表情、智力障碍、走路不稳和极度活跃,有些患者可能还会患有癫痫、喂食困难和睡眠问题。
目前针对angelman综合症没有治愈的方法,临床治疗都是对症缓解,包括癫痫患者使用抗癫痫治疗,步伐失衡患者考虑理疗,智力障碍患者考虑特殊教育等。因此,对于有UBE3A基因缺陷的家庭来说,在备孕阶段考虑做泰国试管婴儿技术(PGD)是一个合理的选择方案。
angelman综合症症状
angelman综合症的典型症状通常包括:
智力障碍,包括运动迟缓和言语迟缓(讲话吐字非常非常的慢),脑电波可以显示异常的构造
共济失调,包括走路失衡和同手同脚走路
特殊面容,包括奇特的笑容和开朗的性格,通常伴有小头症状(比同龄人脑袋更小些)
癫痫
喂养困难
睡眠困扰,以及注意力难以集中
angelman综合症的具体症状因人而异,例如,一些患者可能会癫痫发作,而另一些人可能不会。大多数人患者存在发育迟缓和学习障碍的问题,由于只能缓慢说几个有限的字,导致他们的沟通和交流存在巨大障碍。
早期患者可能会出现共济失调,例如走路时失衡,甚至是同手同脚走路,远看上去就像木偶人在走路一样。部分患者可能还会出现癫痫发作,导致其走路姿势更为缓慢和僵硬。大多数患者都有一种奇特的愉悦表情,经常出现不恰当的大笑和无端微笑。部分患者患有小头畸形,这可能和婴儿阶段的喂养困难有关。
睡眠障碍对于这类患者来说非常常见,主要是对于光线、声音、温度等都异常敏感,这导致了患者通常难以入睡,或者入睡后极易惊醒,这也间接导致了注意力不集中的现象。
angelman综合症病因
angelman综合症的病因已经明确,就是15号染色体上的UBE3A基因发生缺陷所致。虽然目前已经确认这种疾病是常染色体显性遗传疾病,但目前临床所见,约70%的患者是遗传自母亲,约20%的患者是遗传自父亲,另有10%患者是新发突变。
目前已知,UBE3A基因作为一种泛素蛋白连接酶,控制大脑的发育和脑神经递质的活跃,位于15号染色体q11-13区域的约4Mb大小的缺失。在这一区域内,除了UBE3A基因外,还有OCA2基因(控制皮肤、头发和眼睛的色素沉着)和患者的浅色皮肤与白色头发有关。
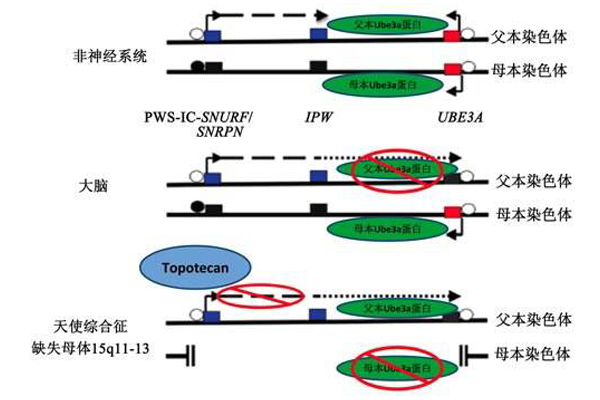
angelman综合症遗传模式
安琪尔曼综合症是一种常染色体显性遗传疾病,这意味着父母中有一方患病,则其后代患病几率为50%。但实际临床中发现,约70%的患者是遗传自母亲的UBE3A基因缺失,约20%的患者是遗传自父亲的UBE3A基因缺失。因此对于有UBE3A基因缺失的家庭来说,最好还是在备孕阶段考虑试管婴儿治疗更好些,尤其是对于那些头胎是angelman综合症患儿或有angelman综合症家族史的家庭来说。
angelman综合症治疗
目前还没有治愈angelman综合症的方法,但可以通过各种维持治疗来缓解症状,不过缺点是需要终生维持治疗,例如:
有些患者有喂养困难,可以考虑用吸管和特制的营养套餐
对于有共济失调和智力障碍的患者,可以通过理疗和特殊教育来改善
如果出现癫痫发作,可以考虑抗癫痫药物
睡眠问题可以通过少量服用安眠药或改善睡眠环境来解决
通常来说,患有angelman综合症的患者,其寿命和正常人无异,但这类患者通常无法孤立生活或工作,需要在集体环境中得到更多的照顾和支持才能生存。但这非常麻烦和耗时耗力,通常建议对那些患有angelman综合症的家庭进行遗传咨询。
angelman综合症遗传咨询
如果有angelman综合症的家族史,或者已经生下一个患有这种疾病的孩子,那么必须要接受遗传咨询,以便规划未来的怀孕。遗传咨询是向个人和家庭提供关于遗传疾病的性质,遗传和影响的信息,以帮助他们做出明智的医疗和生育决定的步骤,一般需要遗传学家调查家族史及遗传病史情况后,进行遗传风险评估。由于angelman综合症以常染色体显性方式遗传,因此父母任意一方有UBE3A基因缺失,其后代就有50%几率遗传,一般推荐使用泰国试管婴儿技术(PGD)进行干预。
PGD又叫胚胎植入前筛查,现在也有叫植入前基因检测(PGT-M),是一种检查胚胎基因或染色体异常的基因技术,经过测试的胚胎如果没有胚胎异常(如没有UBE3A基因异常),将放回子宫继续发育。从临床上看,胚胎通常是在出生后2-6天植入子宫的(即新鲜胚胎移植),但现在,更常见的是冷冻胚胎,以便让女性有更多的时间调理身体,方便后续植入。
这一技术和常规的试管婴儿技术(IVF)非常类似,所不同的是,PGD会对植入前的胚胎做进一步的基因检测,从中排除掉有染色体异常或遗传病的胚胎,从而保证植入子宫体内的胚胎不会患有angelman综合症。
