药审改革五年考,这场改革应该被国人记住

2020年8月18日,一个平平无奇的周三,一周中工作最忙碌的日子,鲜有人特别注意。
17:10分,国家药品监管管理局直属行业报纸中国医药报官方微信上发了一篇文章《药审改革五周年成绩单》,大红的底色为五周年祝福。
少数人转发到了朋友圈,更多人在一天的忙碌中有意无意地忽略了这条波澜不惊的文章,医药圈内外已难寻五年前改革大幕拉开时的盛况。
2015年8月18日,国务院发布《关于改革药品医疗器械审评审批制度的意见》(以下简称《意见》),标志着药品审评审批制度改革(以下简称药审改革)正式开启。
改革开启前,除了浩瀚的改革文件、强力的执行信念,各界无不夹杂着几分怀疑几分兴奋的复杂情绪。
五年来,中国的药监部门度过了最忙碌的五年:从打击数据造假,解决审评积压入手,再到提高仿制药和新药标准,简化审评程序,增加审评力量,鼓励创新,一步步走向国际化。
这是一盘划时代的大棋,结局未定,但数据足以证明过去五年的改革是成功、至少是极具成效的。
审评积压的的现象大大扭转: 截至2019年底,排队等待审评的注册申请已由2015年9月高峰时的近22000件降至4423件。
新药审批数量不断上升: 2016年药审中心共受理化药创新药注册申请90个品种,较2015年增长18%;2017年上升为149个,较2016年增长了66%。2018年受理1类创新药注册申请264个,较2017年增长了21%;2019年上升为319个,较2018年增长了20.8%。
仿制药一致性评价:意见要求提高药品审批标准,将仿制药由此前的“仿已有国家标准的药品”调整为“仿与原研药品质量和疗效一致的药品”。截至2020年1月,国家药监局已发布24批仿制药参比制剂目录,共计2446个药品规格。
制定上市药品目录集:2017年12月29日,CDE位点上线启动《中国上市药品目录集》,通过仿制药质量和疗效一致性评价的药品均纳入《中国上市药品目录集》。
上市许可人制度:截至2019年7月底,10个试点省(市)药品注册申请人共提出持有人注册申请2437件——在不到一年时间里,持有人注册申请增量超过了前三年的总量。其中,有不少是具有自主知识产权、尚未在国内外上市的1类新药。
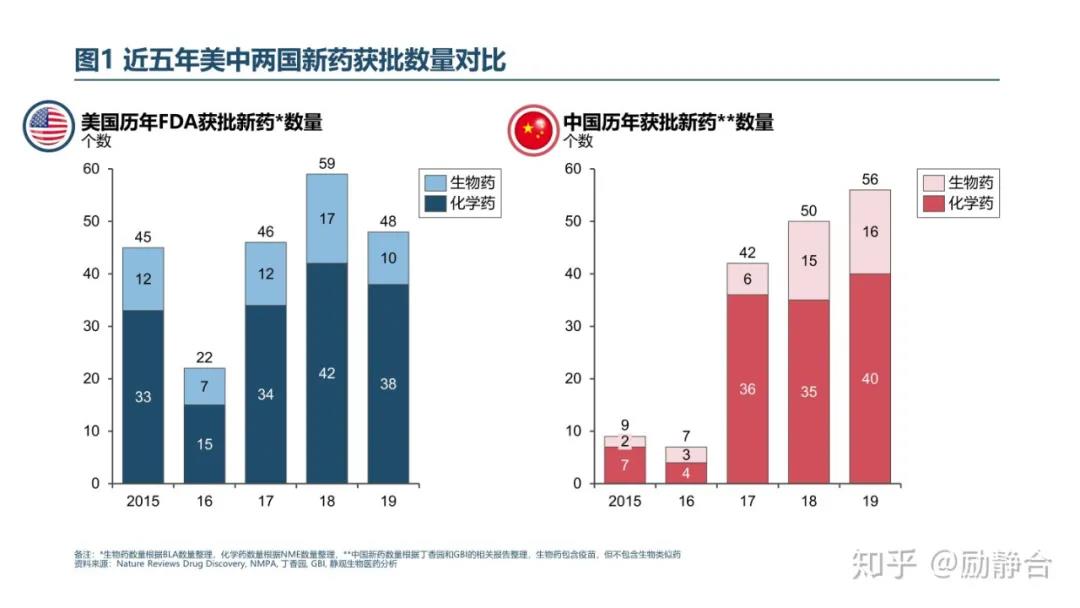
新药获批:自从2016年国家开始加速新药审批速度以来,每年都能获批40个以上新药。2019年首次在中国获批的新药有60个,包括4个中药和56个西药,西药中包含43个进口药和13个国产药。不久前,有人总结了中美两国新药获批数量的对比如下:2019年国内新批准新药(化药)数量一共56个(含首仿),中国的新药获批数量超越了美国。
获批速度:2018年来,进口药在中国首次获批时间比国际(美国、欧盟、日本)获批时间平均晚6.25年(中位4.2年),而2019年的平均滞后时间为5.55年。
产品种类:与2018年相比,2019年获批上市的药物开始向更广阔的治疗领域铺开,主要体现在改变治疗模式(如PD-1免疫抑制剂、银屑病用药)、药品更新换代(如COPD)等方面.
诸如此类,不胜枚举。
这些年来,由于体制不顺的障碍、机制创新的缺失、行政体制的弊端,让社会和行业对目前药品监管有诸多“诟病”,终于被一场自上而下的药审改革打破了多年的沉闷和迟滞。
2015年,沉睡已久的医药产业冰河解冻,大地回暖。从几百份文件次第出炉,到临床数据的断腕自查,再到激发新药创新大幕拉开。上海市食品药品安全研究会会长、上海市食药监局原副局长唐民皓看到国务院44号文后感叹,“感慨和欣喜交织。”
2017年1月12日,食药总局公布目前我国药品注册申请的积压项目已由2015年高峰时的近22000件降至2016年底的8863件。而化学药和疫苗临床试验申请、中药民族药各类注册申请已实现按时限审评。146个具有明显临床价值的创新药、临床急需药、专利过期药和国内首仿药,实施了优先审评。
举例而言,某大型跨国企业在2017年初提交了新药申请。该药适用于严重的肿瘤疾病,具备优先审评资格。经过快速审批后,于2017年3月获得批准,一个月后就进入了市场。中国本土药品也从优先审评通道中获益。2017年,本土研发的埃博拉疫苗仅用了十天时间就获得了新药申请的审 批,但获批条件是保证后期完成第三阶段试验。2018年,9价HPV疫苗从4月20日提交上市申请到4月28日正式获批,只用了8天。
五年间,这样的例子不胜枚举。
彼时,从美国FDA刚回国半年的何如意博士惊叹于这场改革的速度和力度——“比美国的改革快太多了。”
01
十年慢车道
15年来,中国药品审批经历了数次过山车式变革。
从首任国家食品药品监督管理局局长郑筱萸受贿被处死刑的2006年开始,药品审批从“快”字当头走向“大塞车”。在近十年,“药品审批滞后”的舆论批评不绝于耳。
据业内人士统计,2007年到2009年是恢复期,基本没有新药批出;2009年到2013年,化学药品(包括新药和仿制药)的批复数量均呈直线下降趋势,五年一共批复国产药品文号2663个,仅占到当时市场上文号总量的不到2%。
“那几年,政府的导向是少批药,谨慎再谨慎,企业苦不堪言。”北京一位不愿透露姓名的药企老总说。
“局长都被枪毙了。”食药总局一位官员对南方周末记者坦承压力太大了。这次改革之前,药监系统有些官员甚至连学术交流会都不愿参加,“人家会说,那么多药还没批,还有空来参会?”
过于谨慎的代价要所有人共同承受。
一方面,市场上堆积着低水平重复的国产仿制药,有些稍有医药背景的业内人士会选择不用,因为可能“安全却无效”;另一方面,国外大量新药进不来,非法代购甚嚣尘上。“印度抗癌药代购第一人”陆勇,因协助病友购买印度仿制的治疗白血病药物,卷进司法漩涡。“中国人为什么吃不上新药”的疑问一再被媒体提起。
几乎每年全国两会,都有大量针对药品审批难题的提案。在2015年医药界代表委员座谈会上,就有5位全国人大代表重点提及这一困局,认为“中国人要比国外平均晚8-10年吃上新药”。当时食药总局解释了难点,但大家并不买账。在企业和公众眼里,批得出、用得上好药才是真正的需求。
随后,食药总局对外宣布,计划3年消化药品审批存量,平衡增量。半年之后,这场改革风暴悄然拉开序幕。这也是时任食药总局局长毕井泉在2015年1月上任之后力主进行的。
毕井泉引领的改革初衷很简单,就是希望老百姓吃上安全有效的药物。“这么多年,人们都在关注安全性,因为出了那么多药害事件。但没有人关注有效性,以为我们的药品都是有效的,自己的病都是吃药治好的。”
履历显示,毕井泉自1984年进入国家计委(国家发改委前身)工作。任职期间就曾多次就药品价格问题发表看法,并表示“必须推进药品价格改革”,他的调任也被认为或与药价管理体系改革有关。
“非常有魄力,敢担当。”这是业内外人士对毕井泉的一致评价。
之后,药监改革正式进入正轨,业内外评价药监局像是更新换代了好几代。如果说临床试验自查核查、仿制药一致性评价是为了避免有害或无效的药品轻松上市,那么改变新药定义、开启上市许可人制度、优先审评则是为新药进入市场打开大门。
直到现在仍有人对监管部门两天发布四条突破性提案津津乐道,2017年5月11日和12日,原食药监总局发布四份文件,旨在从加速审批流程、临床试验管理、生命周期管理和保障创新者权益四个角度鼓励创新。1个月之后,中国正式加入国际人用药品注册技术协调会(ICH),与国际接轨。
不是没有阻力,但在毕井泉看来,无论反对意见如何,只要能找准定位,搞清楚道理,有据可循,各方都会服理,监管部门也不能再“揣着明白装糊涂”了。
“这就是公道,就是科学。100号文发布后,企业都不再发牢骚出难题,乖乖回去想办法了。谁也不再心存幻想,谁也不再怀疑监管部门的决心了。”
这些只是铁腕的一面,后来回忆起来,改革者还是不改初衷。
2020年8月18日这天,端端酱小心翼翼地问了当时的主政者,五年期满达到您的目标了吗?
答:“认识都是逐步深化的。2015年主要是从解决积压入手,提高仿制药和新药标准,打击数据造假,简化审评程序,增加审评力量。2016、2017年才真正形成和建立鼓励创新的思路及制度。”
02
疫苗案转折点
两年前的2018年,也是一个八月,山东疫苗事件后,国家公布了长春长生公司问题疫苗案件调查及有关问责情况的汇报,时任国家食药监总局的局长毕井泉引咎辞职,引发业内震荡。
当时我在朋友圈写了一条评论,雪花般的留言表达了大家的震惊:
“药监的悲哀,20年,届届改,年年折腾,有理想有能力的人血都冷了,调走的调走,离开的离开。”
“有太多人需要为疫苗事件负责,而毕局绝对不应该是那个引咎辞职的人。”
“没办法,总有个人为此背锅,毕局的改革方向已经深入人心了。只要没有政策复辟,就算毕老的成功了。”
“政策的连续性对创新企业尤为重要!”
“问责了一个最会做事的。”
“他的悲剧和林则徐有异曲同工之处,作为一个远见卓识的改革者,个人之力无法撼动一个病入膏肓的体制。我们最担心的是他好不容易建立起来的药政改革,恐怕又要倒退好多年了。”
“功劳大家记得就好,都知道他去顶雷了,没办法。”
“毕是改革派,当年,他全力推动中国盐业的改革,但也功亏一篑。直到2017年出了一个不伦不类,人人讨厌的盐业改革方案。”
“明白人都会感激他!历史会铭记他领导的改革。”
“看到这个问责心理太难受了,毕局真的做了很多。”
“毕局是我见过最用心工作的领导,他对中国药品管理的贡献无人能比。”
“公道自在人心。”
“问责必须科学,否则适得其反。”
“一声叹气。”
……
改革主政者的突然离开了这一岗位,遗憾、伤心、担忧的情绪延宕在体制内外——不仅为当事人惋惜,更多疑问集中在“改革还会继续吗?药监会再走回头路吗?接下来到底会怎么走?”
2018年3月,新一轮国务院机构改革方案出台,关于食品药品领域的监管体制进行了彻底改革——不再保留国家食品药品监督管理总局;组建国家药品监督管理局(以下简称国家药监局),由国家市场监督管理总局管理。不少人把这一变革看作是改变难以继续的开始。
在改革主政者离开的好一段时间,整个药监系统像是没了主心骨,大家都眉头紧锁,重要文件出台前,遇到难题时,他们仍会向老领导求助,确保改革仍能稳步前行,遵循初心,而老领导一如往常,耐心认真地提出所思所想,并不时做下笔记供相关部门讨论。
事实上,改革者的锋芒不会因为岗位变动而就此停止。按照中国组织人事的规则,大部分正省部级官员在卸任党政职务“退居二线”时,都会归于全国人大或全国政协。只是因为民族、资历、此前的专长和卸任时机不同,路径亦有区别。而一旦他们进入到人大或政协,面对法律提案或草案中不合实际乃至被诟病的地方,往往是最深有感触和能提出建设性意见的人物。
2018年9月28日,《自然综述:药物发现》上发表了一篇Reforming China's drug regulatory system的评论文章,中国的药监改革得到了国际关注。

核心人物的陡然离开让系统内外一度十分消沉,但已经拉开的改革之弓却无法回头。我曾写过,从某种程度上说,这场改革是一场回归之路。
先是回归制度本身。从原先不严谨不用心的“懈怠”状态,回归到制度执行的本来要求,回归到保证药品质量安全的“应然状态”。
其次是回归科学监管。各界都认为五年来监管部门的法律意识明显增加,在出台各项法规中,他们都会首先查看是否会与现有法条冲突。如果有冲突,即请示全国人大授权解决。
药审改革第四年,《药品管理法》迎来近20年来最全面的一次修订,南开大学法学院教授宋华琳认为此次修订“将审评制度改革经验以法律形式固定了下来正在修订当中的《药品专利法》 也有望解决此前审批时间流程过长,新药在我国上市后剩余专利期太短,导致生产企业在研发创新上的积极性不高的问题。”
尽管这次“大修”受到的褒扬不一,不少希望能固定下来的改革条目并没有落实在法律框架之下,但近20年来的全面修订并最终发布已是极为难得。
03
疫情之下新挑战
2020年新冠席卷全球,公众对药品和疫苗的关注度升至最高。药物临床试验也经历了改革后的首次大考。
在湖北,为了找到最有效的药物和治疗方案,新冠期间的临床试验一度出现混乱。
“(武汉)一个病人要参加五个临床试验。这样的临床研究能出什么样的结果?”某国内知名肿瘤科教授为这样的场景担忧,在他看来,临床试验陷入困难不仅是疫情带来的突发状况,更要清醒地认识到,中国临床研究的水平和先进发达国家还有很大的差距。
此前,到处找不到临床试验机构的现象,随着机构认定的放宽而逐渐解决。2017年1月12日,国务院发文取消了临床试验机构的省级初审政策,但只有数量是不够的。
2020年3月3日,上海交通大学第六人民医院朱长太团队联合南京中医药大学,山西医科大学,浙江大学等多单位在medRxiv上发表未经同行评审的研究论文“Systematic Review of the Registered Clinical Trials of Coronavirus Diseases 2019(COVID-19)”,该研究检索了中国临床注册中心和ClinicalTrials.gov的数据库,共获得75项COVID-19注册的临床试验(63项干预性研究和12项观察性研究)。
这是对中国新冠疫情期间已注册临床试验进行的首次系统回顾,文章结论却不容乐观:各种类型的临床试验秩序混乱且数量激增,由于试验质量差、样本量小、完成时间长,在今后相当长的一段时间内,我们将无法获得可靠、高质量的临床依据。
文章提到,部分临床试验缺乏科学设计,将导致质量不高。例如,所有63个介入型试验均未澄清失访偏倚,即观察病例退出研究队列,部分介入型试验在随机性上有很高的偏倚风险。其中,有17个试验没有提及随机性,而有6个试验被判定为非随机试验;很少有试验进行分布隐藏;只有9个试验对参与者、人员和结果评估实施了盲法,以减少试验主观因素的影响。
此外,多数观察试验在评估结果、结果随访和对研究队列随访的充分性方面都有很高的风险偏倚。因此,文章得出结论,无论是介入型试验还是观察型试验,方法论质量都不高。
还有学者直接评价道:到目前为止,中国的疫情已经基本结束,中国的临床试验也已经基本结题,但似乎并没有出现什么值得欢呼的好消息,这些证明药物疗效的临床试验,可以判定近乎全都“翻车”了。
不仅如此,疫情之下新冠之外的临床试验被陡然按下了暂停键。
特殊时期如何做好临床试验,已有的试验和新开始试验如何协调,中断的数据能否再用,已入组和未入组的病人如何安排,病人安全、试验质量如何保证,没有人给出答案。
2020年5月8日,北京大学肿瘤医院副院长沈琳联合多位专家、药企研发成员专门开会讨论“疫情下药物临床试验困局如何破解”。
会上,一个关键问题便是,我们的药监部门为何没能早些出台疫情下临床试验如何进行的指导意见?
北京一位大三甲医院临床试验负责人每天只能通过朋友圈了解各种各样的地方机构/医院/药企在公布各自的应对措施,“五花八门”。
新冠疫情流行期间,FDA(美国食药品监督局)和EMA(欧洲药品管理局)分别于2020年3月18号和3月20日各自发布了“在COVID-19大流行期间的临床试验应对指南”。
对比欧盟和美国两份指南后发现,两者都保持了高度的一致,比如对于方案修改以及记录、受试者的知情权和安全、福利等。尤其是两者有着共同的目的-要严格遵守“三原则”:
(1)维护试验参与者的安全
(2)研究数据的完整性以及
(3)GCP的贯彻执行
对于正在开展的临床试验,FDA和EMA的指南中都很明确地共同指出:
(1)线下的访视流程尽量转移到线上,以电话/视频等形式进行。
(2)根据具体情况,评估是否暂停/停止部分试验中心的临床试验工作。
(3)根据实际情况,暂停或者减缓新受试者的招募,亦或者延长试验周期。
(4)试验的实验室检查等等检查,可以根据实际情况在当地的医疗机构中进行,不一定要送到试验中心进行。
(5)未开始的临床试验,如果是与COVID-19无关的,可以考虑推迟启动。
中国的不少医院和机构的指南也都借鉴了上述两份文件。
4月30日,中国的药监部门终于出台了类似政策——《新冠肺炎疫情期间药物临床试验管理指导原则》征求意见稿,但该原则直到7月14日才正式实施,此时国内疫情主战场已基本结束,各大临床试验也已有序恢复。
也许还能更快更完善一些,但只要走在了正确的道路上,也可算是功不唐捐了。
在全球,新药研发的进度都已肉眼可见的速度不断增长。2019年9月,Insight全新新药数据库显示,全球范围在研的新药超过3万个,仅癌症领域就超过1万个。
中国医药研发的热情只增不减。
2020年7月10日,同写意论坛在苏州金鸡湖召开数千人的大会,这是自新冠疫情以来医药领域首个线下活动,据主办方介绍,参与者多达两千人。一位参会现场的医药人在朋友圈写道,在会场里一天走了一万多步,仍不过瘾。
“中国生物医药产业已经进入新的黄金发展期,各利好政策持续引导产业发展、刺激资本投入、加速人才汇聚,作为中国药企创新发展提供了肥沃土壤”。上述会议总结道,国家创新政策的驱动下,已有越来越多的资金聚集到生命科学领域,显著催生着创新药开发比重,而“本土新药研发企业逐步具备国际药企比肩的能力——攀向更高峰只是时间问题”。
无论上述论断是否过于乐观,这场改革及所有为之付出的人们都应该被记住。一个共识再次被重申:如果医药行业的每一次改革都能为普通民众的健康带来获益才算是真正的成功。
