地中海贫血能活多久
群里的朋友常问起地中海贫血能活多久, 一般轻型地中海贫血不会有生命危险,几乎没有任何症状。中型地海贫血有轻度到中度贫血症状,不会有生命危险。
重型β地中海贫血患者在出生时无症状,几个月的时候发病,患者出现脸色苍白等贫血症状、发育迟缓、易感染。如果不进行治疗,可出现骨髓扩张,使骨骼更宽,更薄,更脆,骨骼更容易破裂,并可能导致骨骼结构异常,特别是在面部和头骨的骨骼,脾脏变大,器官衰竭等症状,最终因重度贫血和器官衰竭而亡,没有治疗的重型β地中海贫血患者一般活不过5岁。
重度α地中海贫血一般在30-40周中在宫内死亡,或者出生数小时后死亡,胎儿出现全身水肿、腹水、胸水、黄疸和重度贫血。
如果接受规范治疗,在现有的案例中,重型β地中海贫血可活到50多岁。接受干细胞移植是治愈地中海贫血的唯一方法,移植成功后,患者不再依赖输血治疗,可有正常人的寿命。造血干细胞移植可用于治疗几十种疾病,例如:白血病、淋巴瘤、多发性骨髓瘤、某些恶性实体肿瘤、再生障碍性贫血、重症免疫缺陷病、急性放射病、重型地中海贫血等。
----
地中海贫血分型
按照血红蛋白(hemoglobin, Hb) 的4级结构, 每个Hb分子由2对珠蛋白肽链构成的球形4聚体, 一对是α链(α链和ξ链) , 另一对是β链(ε、β、γ、δ链) 。这6种不同的珠蛋白链组成了人类不同的6种Hb, 分别是Hb GowerⅠ、Hb GowerⅡ、Hb Portland、Hb F、Hb A、Hb A2, 随发育的不同阶段先后出现, 到成人主要是后3种, Hb A占95%, Hb A2占2.0%~3.5%, Hb F小于1.5%。
无论是国际上还是我国, 目前仍根据珠蛋白链缺乏的类型进行地中海贫血命名和分型。α珠蛋白链缺乏者为α地中海贫血(α珠蛋白生成障碍性贫血) , β珠蛋白链缺乏者为β地中海贫血(β珠蛋白生成障碍性贫血) , 其他少见的还有γ地中海贫血、δβ地中海贫血(δβ0、δβ+、血红蛋白Lepore综合征δβ融合基因) 和遗传性胎儿血红蛋白持续存在征(HPFH, 多见于黑人, 分为缺失型和非缺失型, Hb F升高, 红细胞内Hb F呈均匀分布, 无明显贫血表现) 等。
α地中海贫血和β地中海贫血又可根据所缺乏的珠蛋白基因的种类、程度及临床表现再分型。α地中海贫血基因位于16号染色体短臂13区3带(16p13.3) , α珠蛋白链完全不能合成者称为α0珠蛋白生成障碍性贫血, 尚能合成少量者称α+珠蛋白生成障碍性贫血;根据α基因缺失数目(决定临床表现的有无和轻重) 分为4种类型, 即静止型(1个α基因异常) 、标准型(2个α基因异常) 、Hb H病(3个α基因异常) 和重型(4个α基因异常, Hb Bart′胎儿水肿综合征) 。β地中海贫血基因位于11号染色体短臂1区2带(11p1.2) , β珠蛋白链完全不能合成者称为β0珠蛋白生成障碍性贫血, 尚能合成少量者称β+珠蛋白生成障碍性贫血;根据临床表现可分为轻型(杂合子) 、中间型和重型(纯合子) 。
----
地中海贫血的诊断
根据典型的临床表现和实验室检查特点, 结合患者的家族史及流行病学, 诊断地中海贫血一般并不困难。重点是其中少见类型的发现和基因类型的精确诊断。
一、种族与易发地区
本病以地中海沿岸和东南亚各国较多见, 我国则以华南地区发病率高, 尤其是广东、广西、海南等地区, 湖南、贵州、云南、四川等也是高发区, 而北方很少见。
二、临床表现和家族史
1.α地中海贫血
(1) 静止型:静止型α地中海贫血患者可无任何临床症状和体征, 其父母一方有α地中海贫血。
(2) 标准型:标准型亦称α地中海贫血特征、轻型α地中海贫血。该型患者的临床症状轻, 存在轻度贫血;其父母一方有α地中海贫血。
(3) Hb H病:Hb H病亦称中间型α地中海贫血。该型患者的临床表现多样, 年幼时多无症状, 约半数患者在20岁以后发病。多数患者病情较轻, 主要表现为轻、中度的贫血和长期慢性溶血导致的肝脾肿大;重者则伴有黄疸;少数可伴骨骼轻微改变, 不影响生长发育, 因此无地中海贫血外貌。妊娠、感染、服用磺胺类或氧化剂药物时可诱发其溶血加重, 某些严重者的表现与纯合子型β地中海贫血类似。往往患者的父母双方都有α地中海贫血。
(4) Hb Bart′s胎儿水肿综合征:一般患有HbBart′s胎儿水肿综合征的胎儿在妊娠30~40周时即可发生宫内死亡, 或在早产或出生后数小时内死亡。患病胎儿皮肤苍白、全身水肿和各浆膜腔积液, 伴或不伴有黄疸、皮肤出血、肝脾肿大, 胎盘大而脆, 脐带水肿明显。其父母均为--/αα型地中海贫血。
2.β地中海贫血
(1) 轻型:轻型β地中海贫血患者大多数无临床表现, 少数可有轻度贫血和脾肿大;其父母一方为β地中海贫血杂合子。
(2) 中间型:中间型β地中海贫血患者可存在中度贫血和脾肿大;少数有骨骼改变(轻度) , 性发育延迟。
(3) 重型(Cooley贫血) :重型β地中海贫血患者出生时正常, 而在6~12个月后其贫血进行性加重, 伴有黄疸和肝脾大;生长发育迟缓, 精神萎靡, 智力迟钝, 性征不全, 易感染;骨皮质变薄, 骨骼变脆, 甚至发生病理性骨折。其中大部分具有典型的地中海贫血特殊面容, 表现为额部隆起、鼻梁凹陷、眼距增宽。患者的父母双方均为β地中海贫血杂合子。
三、一般实验室检查
1.血常规:红细胞呈典型的小细胞低色素性(平均红细胞容积、平均红细胞Hb含量降低) 改变是地中海贫血的特征, 可有或无Hb水平的降低。许多地中海贫血患者均在体检血常规检查中被发现和诊断。不同类型地中海贫血患者的红细胞体积略有差异, 往往随着其临床表现的加重, 平均红细胞容积、平均红细胞Hb含量降低程度也更明显, 重型β地中海贫血的平均红细胞容积常50~70 fl/细胞, 网织红细胞增高可达60%以上。
2.血细胞形态:地中海贫血患者的血细胞形态无特异性, 表现为继发溶血的血细胞形态改变, 如靶形红细胞、“马铃薯片形”细胞。Hb H病患者的血涂片经煌焦油蓝染色后, 可见红细胞中含有灰蓝色、均匀、圆形的颗粒状Hb H包涵体。其骨髓象主要表现为红系增生活跃, 重者的铁染色可示含铁血黄素显著增多。
3.生物化学检测:根据患者溶血的情况, 其血清间接胆红素和血清铁蛋白均有不同程度的升高, 后者也是鉴别缺铁性贫血的主要指标。
四、特异性实验室检查
1.红细胞渗透脆性实验:地中海贫血患者的红细胞渗透脆性降低。由于其红细胞膜存在形态、生化、代谢异常, 在低渗盐溶液中易膨胀破裂而溶血。该试验是地中海贫血群体筛查最简便的方法。
2.抗碱血红蛋白测定:抗碱血红蛋白测定检测的是抗碱Hb, 为Hb F的筛查试验。然而除Hb F外, Hb Barts和部分Hb H也具有抗碱能力, 需通过血红蛋白电泳分析来鉴别。
3.Hb电泳:Hb电泳简单易行, 是临床诊断地中海贫血最常用的方法。不同的Hb等电点不同, 在一定p H缓冲液中带有不同的电荷。当缓冲液的p H大于该Hb的等电点时, 其带负电荷, 电泳时向阳极游动;反之, Hb带正电荷向阴极游动。经一定电压和时间的电泳后, 电荷不同、相对分子量不同的Hb, 泳动方向和速度不同, 从而分离出各自的区带。正常成人Hb在p H为8.6的条件下, 电泳显示3个条带, 即Hb A(α2/β2, 占97%) 、Hb A2(α2/δ2, 占2%~3%) 和Hb F(α2/γ2, 占1%) ;而发生β珠蛋白生成障碍时, 往往Hb A2>3.5%、Hb F>5%。关于地中海贫血的不同Hb定量值总结见表1、2。

不同类型α地中海贫血的Hb
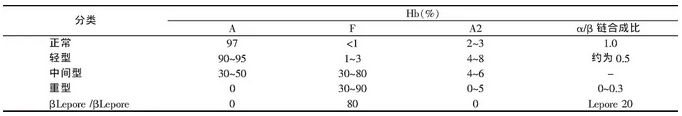
不同类型β地中海贫血的Hb
----
检测方法
Hb电泳
一、醋酸纤维薄膜电泳
醋酸纤维薄膜电泳是临床常用的筛查地中海贫血的方法。醋酸纤维薄膜是均匀的微孔物质, 其对蛋白质的吸附极少, 因此该方法的优点是拖尾现象少、图像清晰、分离速度快、样品用量少且易于洗脱定量。行醋酸纤维薄膜电泳时常规采用p H 8.6的TEB缓冲液, 而采用p H 6.5的TEB缓冲液进行Hb电泳时易分离Hb H与Hb Barts。但醋酸纤维薄膜电泳影响因素较多, 如电压、电泳时间、醋酸纤膜质量、染料浓度等;且由于Hb F与Hb A等电点接近, 很难分辨出Hb A稍后的Hb F区带, 因此醋酸纤维素膜电泳对Hb F分辨率有一定的局限性, 从而影响了结果的可靠性。
二、聚丙烯酰胺凝胶电泳
聚丙烯酰胺凝胶电泳的分辨率高, 可检出醋酸纤维素膜电泳中与Hb A不易区分的不稳定Hb、潜在的异常Hb和绝大多数α地中海贫血, 明确区分β0地中海贫血与β+地中海贫血。本方法简单易行, 对地中海贫血(珠蛋白生成障碍性贫血) 的诊断有重要参考价值。然而, 琼脂糖凝胶电泳不能区分Hb E与Hb A2。
三、毛细管电泳
毛细管电泳综合了电泳和色谱两者技术的优点。与上述传统的薄膜或凝胶电泳相比, 不仅无需染色, 且速度快、所需样本量少。我院实验室采用该技术可以在30 min内一次性将5种珠蛋白肽链完全、准确地分离, 得出Hb A、Hb A2、Hb F、Hb H、HbBarts各组分的百分比。该方法操作简便, 重复性好, 省时高效, 适用于血红蛋白病的快速诊断, 也是目前我国地中海贫血筛查常用的方法。
尽管血红蛋白电泳在技术上不断改进和完善, 但目前仍不能将Hb E、Hb A2两者区分开, 因此无法完全鉴别出部分静止型地中海贫血, 有一定的漏诊率, 需进行基因分析等来诊断这部分人群。
高效液相层析法
国际地中海贫血协会广泛推荐使用HPLC作为Hb分析参比方法。其原理是利用离子交换树脂作为固定相, 根据各Hb的理化性质不同, 其在分离柱中停留的时间也不同, 从而将各Hb分离。HPLC可分离和定量Hb F和Hb A2, 有助于β地中海贫血、δβ地中海贫血、遗传性胎儿Hb持续存在征的诊断。而且HPLC不受标本质量如高脂、黄疸等血标本的干扰, 检测准确率高, 重复性好, 对地中海贫血尤其是β地中海贫血的诊断具有很好的临床价值, 而其不足之处是对α地中海贫血筛查仍有一定局限性, 需进行基因检测以提高诊断的准确率。
质谱分析法
质谱分析法是近代发展起来的快速、微量、精确测定相对分子质量的方法, 在此基础上设计的电喷雾电离质谱和基质辅助激光解吸电离飞行时间质谱, 可提高异常Hb的检出, 作为Hb电泳和HPLC方法的补充。但该方法对大分子物质的解析度有限, 在检测前需用胰蛋白酶预处理成较小的蛋白分子方能进行, 因此临床上仍有部分变异的Hb不能被发现。
基因检测
基因检测是地中海贫血的确诊实验, 具有更直接、特异、灵敏的特点, 可明确基因型及基因缺陷部位, 但实际中不可能对每一个患者都进行全部基因序列的检测。由于大多数的α地中海贫血均由基因缺失所致, 而90%以上的β地中海贫血是由基因突变所致, 所以在基因分析时, 对于α地中海贫血, 先检测常见的大片段缺失, 然后再检测少见的点突变;对于β地中海贫血, 先检测常见的17种突变, 然后再检测少见的大片段缺失。常用的基因检测方法有基因探针、DNA微阵列、限制性内切酶图谱分析、聚合酶链反应(PCR) 、扩增不应突变系统技术、多重突变引物延伸技术、反向斑点杂交、特异性寡核苷酸杂交等一系列分子生物学技术。
一、Gap-PCR
Gap-PCR可用于α地中海贫血基因携带者的一线筛查。基于Gap-PCR原理设计, 建立检测常见缺失型α地中海贫血突变(-SEA、-A3.7和-A4.2) 的单管多重PCR技术, 可成功检出这3种常见缺失型α地中海贫血突变的纯合子、杂合子和双重杂合子, 具有简单、快速、准确、可靠、经济、实用的优点, 目前也常可用于地中海贫血的产前诊断。与常规血液学筛查方法比较, Gap-PCR分子筛查技术更具有特异性和可靠性。
二、PCR-反向斑点杂交(PCR-RDB)
反向斑点杂交法是目前国内对β地中海贫血诊断率最高的方法, 其具有快速、简便、高灵敏度和特异性强的特点。但在反向斑点杂交检测过程中, 斑点信号可能会存在不均一现象, 且该检测受到各种试验因素、条件的影响, 其稳定性和重复性有待提高。
三、基因芯片
基因芯片的检测原理是, 将许多特定的寡核苷酸片段(或c DNA片段) 作为靶基因, 有规律地排列固定于支持物上;样品DNA通过PCR扩增掺入荧光标记分子或放射性核素作为探针, 然后按碱基配对原理将两者进行杂交;再通过荧光或核素检测系统对芯片进行扫描, 由计算机系统对每个探针上的信号作出比较和检测, 从而得出所需要的信息。基因芯片检测的突出优点是高通量, α地中海贫血和β地中海贫血的诊断可集成于1张芯片上进行。地中海贫血的诊断基因芯片将我国人群中常见的3种缺失型α珠蛋白基因突变(特别是对Hb H病) 及β珠蛋白基因突变探针固定于同一张基因芯片表面, 简便、省时, 且无需接触放射性核素, 可同时、快速、准确地检测α与β地贫基因突变, 为临床提供了一种快速、有效的地中海贫血基因诊断及产前诊断方法, 便于推广应用。
四、最新研究
有文献报道, 将多重PCR与磁珠悬浮阵列技术相结合, 操作时间短, 可一次检测100个样本, 同时检测出地中海贫血基因缺失及点突变。相对于传统检测方法, 该技术既方便又可减少花费, 有待进一步推广。lin等利用基于实时荧光PCR的探针高分辨率熔点曲线分析技术, 在40 min完成对常见12种β地中海贫血基因突变的检测, 具有准确、快速、高效的特点, 可作为新生儿血液筛查的项目。
产前基因诊断
因地中海贫血为常染色体隐性遗传性疾病, 在我国地中海贫血高发地区中应推广专项婚前检查。如夫妇双方均为地中海贫血基因携带者, 则应于妊娠期进行产前诊断。可于妊娠第8~12孕周吸取绒毛, 妊娠第16~20孕周抽羊水分离脱落细胞, 妊娠第20孕周后抽取脐带血, 提取胎儿的DNA, 按上述基因诊断方法(单管多重PCR技术、PCR-反向斑点杂交等) 检测胎儿是否获得了地中海贫血的缺陷基因。
近年来, 无创产前诊断技术发展迅速, 其中以胎儿游离DNA检测技术的应用最为广泛。Lin等利用PCR与导流杂交技术相结合, 通过低密度基因芯片对60例孕妇成功进行了常见的3种缺失型及3种突变型α-地中海贫血和17种β地中海贫血基因突变、复合地中海贫血的产前诊断;也有研究通过该方法对141例孕妇成功进行检测。因在同一基因芯片上进行α地中海贫血和β地中海贫血的测定缩短了检测时间, 降低检测成本, 同时减少复合型地中海贫血的漏诊, 适合基层实验室用于常见地中海贫血的诊断。但不足的是, 该方法仅能检测常见类型的α地中海贫血和β地中海贫血, 具有一定局限性, 建议同时进行缺铁性贫血的筛查和地中海贫血基因序列的检测。然而, 干志峰等提出, 胎儿游离DNA研究当中发现的最大问题是母血DNA背景污染的问题, 虽然可根据DNA片段大小不同, 用现有技术加以区分, 然仍不能完全避免。
指趾相对变异量技术在产前诊断中有很好的发展前景, 其主要用于诊断β地中海贫血, 通过检测杂合子孕妇血浆的突变和野生型致病等位基因的剂量, 来决定等位基因是否平衡。若不平衡则为纯合子胎儿, 若平衡则为杂合子胎儿。胎儿DNA越丰富, 指趾相对变异量技术诊断的准确率就越高。Lun等指趾相对变异量技术基础上采用指趾核酸大小选择技术, 可增加胎儿DNA含量, 提高无创产前诊断单基因疾病的可靠性。
植入前基因诊断
植入前基因诊断是针对高危夫妻所进行的、胚胎种植前基因诊断, 也就是第三代“试管婴儿”。通过筛选出没有致病遗传基因的胚胎植入子宫, 可有效防止地中海贫血患儿的出生。主要方法是卵裂球的微活检(从2~8个细胞期的胚胎中分离单个核细胞) 、胚胎冻存和(或) 卵裂球的培养。20世纪末, 中山大学首例植入前基因诊断成功, 现该项技术在我国已被普遍接受应用于α地中海贫血和β地中海贫血高危人群的诊断。
----
α地中海贫血
α地中海贫血是世界上最常见的血液系统遗传病之一, 该疾病是由于α珠蛋白的基因缺失或功能障碍, 导致α珠蛋白肽链合成减少或不合成引起。该病高发于从地中海沿岸的意大利、希腊、马耳他、塞浦路斯到东南亚各国的广大地区。我国南方也是本病的高发地区。
人类的α珠蛋白基因位于第16号染色体短臂末端的α珠蛋白基因簇中, 该基因簇包括2个重复的α基因(α2和α1) 、一个胚胎期类α基因(ζ2) 、三个假基因(ψζ1、ψα2、ψα1) 和一个功能未名的θ1。其排列顺序为5'-ζ2-ψζ1-ψα2-ψα1-α2-α1-θ1-3'。根据分子缺陷可将其分为:基因大片段的缺失型α地中海贫血和少数几个核苷酸的插入与缺失或单个碱基替换的非缺失型α地中海贫血两大类。而就一个单倍体而言, 根据α珠蛋白合成减少的程度, α地中海贫血可为两类缺陷:(1) α地中海贫血1(α°地中海贫血) , 特点为缺失两个α基因(--/) , α珠蛋白肽链合成完全受阻;α°地中海贫血基因具有明显的种族与地理分布特征, 我国最常见的为--SEA, 极少见--THAI, 而--FIL则更罕见;(2) α地中海贫2(α+地中海贫血) , 缺失一个α基因或一个α基因发生点突变或少数几个碱基的缺失或插入引起(-α/、αTα) , 表现为α珠蛋白肽链合成部分受阻。我国最常见的α+地中海贫血基因为-α3.7、-α4.2缺失型突变以及Hb CS、Hb QS、Hb WS点突变。根据临床表现又将其分为静止型(-α/αα、αTα/αα) 、标准型(--/αα、-α/-α、αTα/αTα、-α/αTα) 、血红蛋白H病(--/αTα、--/-α) 与Hb Bart水肿胎儿综合征(--/--) 四类。其中症状最重的为Hb Bart水肿胎儿综合征, 患儿通常不能存活。其次为血红蛋白H病(Hb H) , 其临床表现往往轻重不一, 通常非缺失型Hb H患者的临床表现往往比缺失型Hb H患者更为严重。由于目前对地中海贫血尚无有效的治疗方法, 因此加强遗传咨询, 婚前及产前筛查, 对夫妇双方为地中海贫血基因携带者的孕妇, 在妊娠早期进行产前基因诊断, 防止重型地中海贫血儿出生, 是最有效的预防措施。对于α地中海贫血的基因诊断始于20世纪80年代初, 经历了30多年的发展, 如今对该疾病的诊断越来越准确。本文就缺失型与非缺失型α地中海贫血的基因诊断方法展开综述。

α地中海贫血的分子机制
α地中海贫血是一种减少血红蛋白产生的血液疾病。血红蛋白是红血球中的蛋白质,它携带氧气到整个身体的细胞。在具有α地中海贫血特征的人群中,血红蛋白量的减少阻止足够的氧气到达身体组织。受影响的人会贫血、皮肤苍白、虚弱、疲劳和更严重的并发症。
根据基因类型和严重程度将α地中海贫血分为静止型α地中海贫血、轻型α地中海贫血、HbH病、Hb Bart's胎儿水肿综合征。
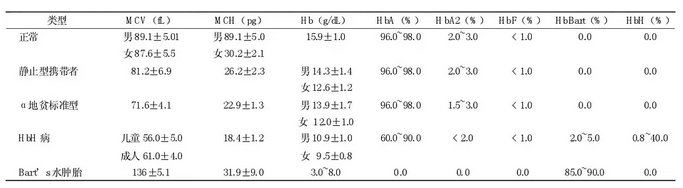
α地贫不同类型的红细胞指数及血红蛋白类型
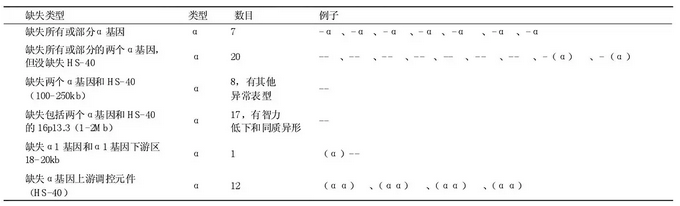
缺失性α地贫的分类
Hb Bart's胎儿水肿综合征是最严重的类型,HbBart综合征的特征是胎儿水肿,出现腹水、胸水。严重的贫血,肝脾肿大(肝脾肿大),心脏缺陷和泌尿系统或生殖器异常。由于这些严重的健康问题,这种情况下的大多数婴儿出生后不久就死亡或死亡。HbBart综合征还可能导致怀孕期间妇女的严重并发症,包括危险的高血压伴肿胀(先兆子痫),早产和异常出血。
HbH病引起轻度至中度贫血,肝脾肿大,以及眼睛和黄疸。一些受影响的人也有骨骼变化,如上颚过度生长,前额异常突出。HbH病的通常在幼儿时期发病,可以活到成年。
静止型α地中海贫血和轻型α地中海贫血症状较轻或者几乎没症状。
α地中海贫血是世界范围内相当常见的血液疾病。每年都有成千上万的Hb Bart综合征和HbH病婴儿出生,特别是在东南亚地区。地中海国家,北非,中东,印度和中亚地区的人们也经常发生α地中海贫血。在我国,地贫主要分布在我国的长江以南地区,广东、广西和海南是高发地区。
α地中海贫血的遗传是复杂的。每个人从每个父母继承两个α-珠蛋白等位基因。如果父母双方都缺少至少一个α珠蛋白等位基因,他们的孩子就有可能患Hb Bart综合征、HbH疾病、轻型静止型α地中海贫血。确切的风险取决于有多少等位基因缺失以及HBA1和HBA2基因的哪个组合受到影响。
一、基于缺失型α地中海贫血的基因诊断方法
1.Southern印迹结合限制性内切酶谱分析:
Southern印迹杂交是研究DNA图谱的基本技术, 在遗传病诊断、DNA图谱分析及PCR产物分析等方面有重要价值, 该方法可用于分析基因的大片段缺失与重复突变。陈坚等利用该技术对一个β珠蛋白基因βIVS-Ⅱ-654(C→T) 杂合子中间型β-地中海贫血先证者家系进行分析, 以Southern杂交技术结合限制性内切酶Bam HⅠ、BglⅡ酶切分析该家系的各个DNA样品。证实先证者为
2.多重PCR结合溶解曲线分析:
该技术运用双链DNA能特异结合荧光染料SBR-GreenⅠ以及不同双链DNA具有不同的解链温度的特点。Munkongdee等运用此技术将PCR引物分别设计在断裂点的侧翼以及断裂点的内部(扩增ψζ和α2) 经单管多重PCR后产物行溶解曲线分析, 结果显示--THIA、--SEA、ψζ与α2的溶解温度分别为(79.9±0.2) ℃、(89.4±0.5) ℃、(92.8±0.2) ℃、(85.0±0.2) ℃, 各地中海贫血基因型特定的溶解温度之间无交叉, 成功的鉴定了各缺失型。该方法具有简捷, 快速的特点, 且由于整个反应过程均是在闭盖的情况下进行, 降低了污染。同时该技术可检测单碱基的突变、小片段插入或者缺失。
3.缺口PCR(Gap-PCR) 结合片段大小分析:
Gap-PCR最早由中国台湾学者Chang等建立。其原理为:设计引物和缺失序列的两侧翼序列互补, 由于α基因的缺失突变使本来正常相距遥远位于断裂点两侧翼较远的引物变得很近, 以至于能扩增出特定长度的片段;另外一对引物则位于缺失区域, 这样在杂合子或完全正常的情况下, 正常等位基因才会扩增出来。根据扩增片段大小此法能很好地区分正常杂合子和纯合子个体, 可用于检测基因的大片段缺失。段山等应用此技术成功的区分了--SEA、-α3.7、-α4.2三种常见缺失突变。Liu等运用此技术分三管多重Gap-PCR成功的检测了--SEA, -(α) 20.5, --MED, --FIL, --THAI, αααanti3.7, -α3.7, -α4.2七种常见的α地中海贫血基因。随着多重PCR的发展, 目前已可以在同一反应管内检测以上八种常见的α地中海贫血基因, 大大地简化了实验步骤。由于该方法只需普通的PCR结合琼脂糖凝胶电泳, 对仪器要求低, 易于推广, 目前被广泛应用于地中海贫血的基因检测。但该方法的显著缺点就是不能检测未知的缺失突变以及非缺失型突变。
4.多重连接酶依赖探针扩增技术(multiplex ligation-dependent probe amplification assay, MLPA) :
MLPA技术最早由荷兰学者Schouten等于2002年提出, 是一种高通量、针对待测核酸中靶序列进行定性和定量分析的技术, 它利用简单的杂交、连接、PCR扩增反应, 在单一反应管内可同时检测40多个不同核苷酸序列的拷贝数变化。该方法具有重复性好, 特异性高等优点, 可同时检测已知与未知的大片段缺失或重复突变, 目前的针对α珠蛋白基因的MLPA试剂框中还添加了针对Hb CS点突变的特异探针, 因此它还能检测Hb CS点突变。Joly等运用MLPA结合CGH-array分析, 发现了一包含整个α珠蛋白基因家族的长达285 kb的大片段缺失。Phylipsen等应用MLPA结合长链PCR与直接测序, 检测出了11种新的缺失型α地中海贫血基因。但由于该方法成本较高, 需要毛细管电泳仪, 从而限制了该方法的推广应用。同时由于该技术能检测罕见与未知的α地中海贫血基因, 因此该方法可作为常见地中海贫血基因筛查阴性但却高度怀疑地中海贫血患者的补充实验。
5.实时荧光定量PCR:
实时荧光定量PCR是目前确定样品中DNA(或c DNA) 拷贝数最敏感、最准确的方法。该方法被广泛应用于DNA基因表达情况的研究, 同时它也可以用于检测基因拷贝数的变化。Fallah等应用此技术对29例经常见缺失型地中海贫血基因与测序检测均阴性的α地中海贫血疑似样本进行检测, 29例样本均检测到了α珠蛋白基因的缺失。该技术同MLPA技术一样能同时检测已知与未知的大片段缺失, 因此它能弥补传统地中海贫血基因检测的缺陷, 提高诊断率。从而能更有效的对地中海贫血进行产前筛查。
二、基于非缺失型α地中海贫血的基因诊断
(一) 针对已知的非缺失型α地中海贫血的基因诊断
1. PCR结合限制性内切酶酶谱分析(PCR-RFLP) :
如果突变产生或消除了一个限制性酶切位点, 用特异的限制酶酶切PCR产物后经电泳分离、溴乙啶染色, 可直接根据观察到的条带的长度差异作出诊断。该方法具有简便、快捷、经济的特点。但这一方法只限于涉及限制性内切酶位点改变的突变和小片段缺失的检测。如Hb CS突变可生成一个MseⅠ酶切位点。Liu等曾用Mse I酶切法检出Hb CS。
2. 多重扩增不应突变系统(multiplex-amplification refractory mutation system, ARMS) :
ARMS原理为针对各突变位点设计相应的引物, 通过扩增突变特异性长度的片段, 然后根据产物片段大小来区分各突变。陈萍等应用该技术, 设计4条引物分别扩增Hb CS及Hb QS及正常α珠蛋白基因片段, 成功的检测Hb CS及Hb QS突变。Lacerra等应用该技术在两PCR反应管内可检测九种已知的α基因点突变。因该技术简便, 快速, 对仪器要求低, 只需简单的PCR扩增与琼脂糖凝胶电泳, 它同Gap-PCR一样, 被广泛地应用于α地中海贫血的基因检测。
3. 聚合酶链反应结合寡核苷酸探针的反向斑点杂交(PCR-RDB) :
反向斑点杂交, 其与传统杂交法区别为:将膜上固定探针取代固定PCR产物, 从而改变了传统杂交法一次只能检测一种突变的不足, 这样在一个杂交反应中能同时分析一份标本中可能存在的多种点突变, 该技术大大提高了诊断效率。郭广洲等应用PCR-RDB针对中国人常见的三种非缺失型α地中海贫血(αQSα, αCSα, αWSα) , 设计针对该三个位点特异性带有生物素标记的引物进行PCR扩增, 扩增产物与固定在膜条上的正常与突变寡核苷酸探针进行杂交, 成功的区分了此三种突变纯合子与杂合子。李莉艳等应用该技术对αQSα、αHSα、αCSα、αWSα、αCD30α、αCD31α、αCD59α六种非缺失型α地中海贫血作出了正确诊断。因该方法具有简便快速准确的优点, 其同ARMS一样被广泛地应用于常见非缺失型地中海贫血基因的检测。需注意的是:由于该方法应用的是探针杂交, 所以探针的特异性以及覆盖范围决定了该方法的灵敏度与特异性。在设计探针时一定要充分考虑探针杂交范围内的多态性位点, 如果突变位点附近存在多态性位点则会使模板DNA不能有效的与膜条上的突变探针杂交, 从而导致假阴性。Yi等就曾报道在β地中海贫血的反向斑点杂交时由于codon 17(A>T) 位点附近的一个多态性位点存在, 而导致将codon 17(A>T) 杂合突变判断为纯合突变的案例。
4. 基因芯片:
基因芯片, 又称DNA微阵列(DNA microarray) , 是把大量已知序列探针集成在同一个基片(如玻片、膜) 上, 经过标记的若干靶核苷酸序列与芯片特定位点上的探针杂交, 通过检测杂交信号, 从而对生物细胞或组织中大量的基因信息进行分析。具有高度并行性、多样性、微型化和自动化等突出优点。Ye等设计了针对常见α、β地中海贫血基因的38个探针(包括β地中海贫血的常见16种突变, -α3.7、-α4.2、--SEA、αQSα、αCSα) , 集α、β地中海贫血常见突变于同一杂交反应, 简化了反应步骤, 能同时诊断α与β地中海贫血。同理Bang-Ce等设计了能同时检测α、β基因的微阵列, 能成功的对α、β地中海贫血进行基因分型。
5. 其他
Southern blotting印迹杂交法、 寡核苷酸(ASO) 探针检测法、突变特异性扩增系统法(ARMS)、 跨越断裂点PCR法(gap-PCR)、 反向点杂交法(RDB)
(二) 针对未知的非缺失型α地中海贫血突变筛查
1. 聚合酶链反应结合单链构象多态性分析(PCR-SSCP) :
SSCP分析是常用的点突变分析技术。在SSCP检测到突变位点存在的基础上, 即可通过DNA序列测定进行验证。赵永忠等运用该技术很好的检测出至少三种与野生型不同的电泳条带。异常条带的样品分别经测序确认为αCS、αQS、αCD122突变。近年随着斑点杂交及测序技术的发展, 该技术已很少被应用于地中海贫血的基因检测。
2. 变性高效液相色谱技术(denaturing high performance liq-uid chromatography, DHPLC) :
该技术可自动检测单碱基替代及小片段核苷酸的插入或缺失, 已被证实其为一种高性价比的检测技术, 敏感性较高, 他能最低检测5%的变异(出现异常峰) 。Liu等利用该技术对92例不同表型的α地中海贫血患者与18例正常人样本进行盲法检测, 发现在分离温度为63.8℃时Hb CS、Hb QS、Hb WS突变表现为不同的峰。而在50℃时--SEA与正常等位基因表现出不同的峰, 该分析结果与患者基因型完全吻合。该方法具有快速、准确、通量高等优点, 适合α地中海贫血的筛查。由于该方法需要专用的高效色谱仪, 同时为疾病的筛查实验, 因此目前未被推广的应用于α地中海贫血的基因检测。
3. 基因测序(gene sequencing) :
基因测序是目前检测基因突变的金标准, 他能诊断已知的与未知的点突变以及小片段核苷酸的缺失与插入突变, 因此较广泛地应用于非缺失型α地中海贫血的基因检测, 目前最常用的是第一代测序技术双脱氧终止法(Sanger) , 由于该检测属于定性实验, 它不能检测α珠蛋白基因的拷贝数变化, 因此它不能检测大片段的缺失。随着测序技术的提高, 出现了新一代测序技术———高通量测序技术, 与传统测序技术相比较, 其一次可对几十万到几百万条DNA分子进行序列测定, 使得对一个物种的转录组和基因组进行细致全貌的分析成为可能。因此其能检测所有类型的α地中海贫血。
随着人们对疾病的深入研究, 发现地中海贫血的临床表现也受一些其他相关基因的影响。如Tufarelli等就曾发现α珠蛋白基因家族下游的LUC7L基因突变造成的反义链RNA存在而导致珠蛋白HBA2基因甲基化失活, 引起一种特殊类型的α地中海贫血。研究还发现HRI基因突变也会影响地中海贫血的表现型。随着新一代测序技术的到来, 相信人们对α地中海贫血的致病基因会有更深入的认识。
随着分子生物诊断技术的不断进步, 使α地中海贫血的诊断更加全面与简捷。但各方法各有利弊, 应根据各方法的适应范围、灵敏度、特异性等综合考虑, 选择方法。
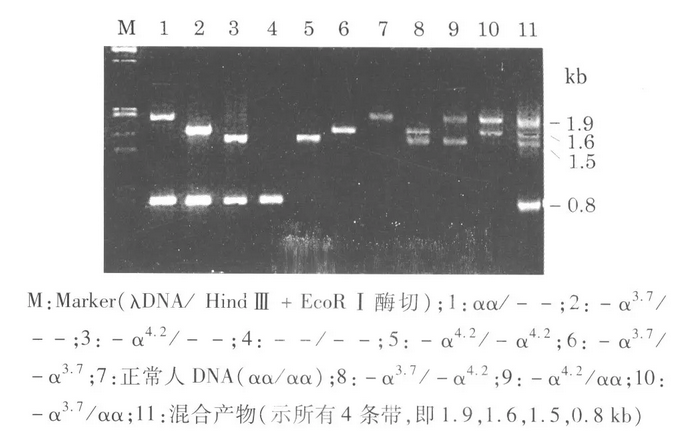
用mPCR检测中国人中10种α地中海贫血基因型的电泳图谱
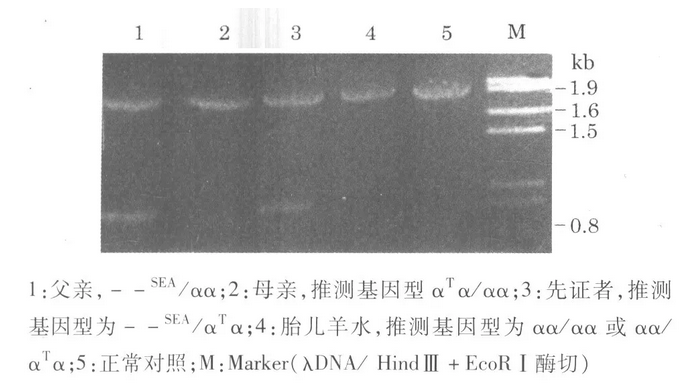
mPCR技术检测α地中海贫血家系成员DNA α地中海贫血基因型的电泳图谱
一般疗法
输血治疗
输血是治疗中间型α地贫的重要方法之一。单纯输红细胞悬液, 使Hb维持在60~70g/L, 虽可延长患儿生命, 但不能改善患儿的生长发育障碍, 且其生存质量随年龄增长越来越差, 多于第2个10年内因脏器功能衰竭而死亡。因此, 低输血疗法(保持Hb>70g/L) 正逐渐被高输血疗法(维持Hb> 100g/L) 和超高输血疗法(维持Hb>140g/L) 所取代。目前提倡的是:先反复输浓缩红细胞, 使患儿Hb含量达120~140g/L, 然后当Hb≤80~90g/L时每隔3~4周输浓缩红细胞10~15ml/kg, 使Hb维持在100g/L以上。高量和超高量输血更利于保证患儿的正常生长发育, 抑制骨髓外造血, 减轻肝脾肿大, 减少肠道吸收, 减轻骨骼畸形和慢性低氧血症, 并减轻心脏负担以延长患儿的生存期。另外, 成分输血正替代输全血。输给由血细胞分离器所采集的主要含年轻红细胞即年幼红细胞的血液, 可延长输血的间隔, 进一步减少肠道铁的吸收, 使铁在体内堆积明显减少。
铁螯合剂治疗
地贫患儿由于长期反复输血必然导致铁负荷过重, 铁质沉积导致器官功能异常, 尤其是损伤心肌导致死亡。为了控制铁的过度负荷, 应用铁螯合剂可延长患儿的生存期。近年发现过早用铁螯合剂可导致患儿铜、锌缺乏, 影响小儿骨生长。亦有研究发现铁螯合剂对小儿骨生长和骨髓腔有直接损害作用, 过早应用可致骨骺端的改变, 故提出最好推迟到3岁以后, 或患儿在接受20次以上输血后并有铁负荷过重的证据, 血清铁水平在800~1000mg/L, 血清转铁蛋白完全饱和时使用为宜。目前临床上常用的铁螯合剂为去铁胺(deferoxamine) 。去铁胺是一种大分子络合物, 以1∶1比例与铁原子结合, 可以增加铁从尿液和粪便中排出, 与VitC合用可使尿中铁的排泄量增加1倍。剂量为20~50mg/(kg·d) , 溶入注射用水或生理盐水, 用便携式输液泵每日(或每晚) 腹壁皮下注射8~12h, 每周连用5~6d。用药前后应做血清铁蛋白(SF) 、尿铁的监测。若SF>3000μg/L或出现铁负荷激发心脏病时, 可予去铁胺50~70mg/(kg·d) 持续24h静滴。由于皮下或静脉应用去铁胺有一定困难, 许多患儿不方便或不能应用, 现已有一种新型口服铁螯合剂去铁酮(deferiprone) 用于临床, 这是一种小分子螯合剂, 以3∶1比例结合铁元素, 对去除心脏铁负荷比去铁胺更有效。
脾切除和脾栓塞治疗
中间型α地贫(Hb<80g/L, 无黄疸) 行脾切除疗效极佳。脾切除后红细胞破坏减轻, 可改善患者的贫血状态, 减少输血量, 术后患者几乎不用输血, 延长生存期。但脾切除后有可能出现继发性免疫功能低下, 并发严重感染。近年来, 大部分脾栓塞法采用50%~85%栓塞。剩余的脾组织保留了足够的免疫能力, 术后体液免疫与细胞免疫降低不明显, 避免了脾切除后的凶险感染;另外由于栓塞后脾已形成包裹不再增生肿大, 避免了部分脾切除后复发脾肿大和脾功能亢进的可能。因此认为大部脾栓塞较脾切除法手术安全、简便、经济, 术后恢复快, 住院时间短, 是目前治疗Hb H病的重要方法之一。
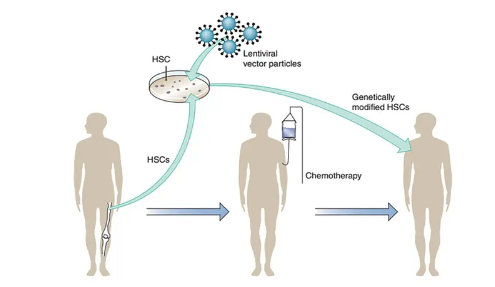
造血干细胞移植
目前认为造血干细胞移植(HSCT) 是根治重型地贫的惟一方法, 包括骨髓移植(BMT) 、脐血移植(UCBT) 、外周血造血干细胞移植(PBSCT) 和宫内造血干细胞移植(IUSCT) 。
BMT的实质是HSCT。干细胞具有自我复制并分化为成熟血细胞与免疫活性细胞的能力。供体可分为血缘相关人类白细胞抗原(HLA) 全相合、血缘相关HLA不全相合(包括同胞、双亲) 或单倍体相合、非血缘相关供体三类。目前任何一种HSCT治疗α地贫主要是血缘相关HLA全相合者, 其中首选BMT/PBSCT, 其次为UCBT。但由于血缘相关HLA 全相合供体来源有限(仅25%~30%患者可找到HLA相合的家系成员供体) 而限制了BMT的开展。PBSCT也同样因 HLA 相合供体来源有限的原因而限制其开展。脐带血中含有丰富的造血多能干细胞和定向祖细胞, 有着更大的潜在分化、增殖能力, 具有增殖快、可以扩增、集落形成高、来源广泛等优点, 因此, 脐血干细胞以其独特的生物学特性、资源优势以及临床适应证的广泛性等可以弥补BMT的不足。脐血中Hb、红细胞含量比成年人高30%~50%, 且胎儿Hb对氧的亲和力强, 用来治疗贫血最为理想。另外UCBT最大的优点是能部分克服 HLA 不全相合的障碍, 移植物抗宿主病发生率及程度较BMT低。若年龄小, 输血量少, 则UCBT成功的机会更大。
Sodani等发现, 年龄小于17岁者接受HLA相合的供体移植前给予白消安(busulfan) 14mg/kg和环磷酰胺120~160mg/kg, 移植后地贫复发率为30%。这种结果可能与免疫抑制缺陷或对地贫骨髓的根除不充分有关, 或两种原因同时存在。为了增强免疫抑制及对地贫集落的根除, 在使用白消安和环磷酰胺的同时使用羟基脲、硫唑嘌呤和氟达拉滨(fludarabine) , 连续有33例小于17岁的患者采用了这种用药方法后, 生存率达93%, 且移植后地贫的复发率降至了8%。
重型α地贫子宫内治疗是有必要的。对于严重的Hb Bart's胎儿水肿综合征, 一经产前确诊即可对胎儿进行换血治疗, 保住胎儿性命, 出生后适时行HSCT治疗可以提高患儿的生存率。但目前IUSCT成功所需单个有核细胞数、移植的最佳胎龄、植入后的状态尚待进一步深入研究。
基因治疗
基因治疗是指运用基因转移技术将遗传物质导入生殖细胞或体细胞, 治疗基因缺失、突变等异常所引起的遗传性以及遗传相关的疾病。地贫作为一种单基因缺陷的遗传病, 理论上是基因治疗的理想模型, 但由于珠蛋白基因在胚胎至成年人的发育过程中表达调控的复杂性, 真正的转基因治疗仍处于动物实验阶段。近年来以小鼠为实验模型研究β地贫的基因治疗取得了很大进展, 但α地贫的相关基因治疗还需进一步研究。
----
β地中海贫血
β地中海贫血因为基因异常,造成β-蛋白肽链的合成受部分或完全抑制,导致正常血红蛋白数量减少以及过剩的α-珠蛋白肽链聚合形成异常血红蛋白。使得血红蛋白的携氧能力下降,凋亡速度加快,无效造血, 血管外溶血增加。
不论是β地中海贫血还是α地中海贫血,都可以根据症状的严重程度分为重型地中海贫血、中间型地中海贫血和轻型地中海贫血。轻度地中海贫血(地中海贫血性状)的个体通常有轻微的无症状红细胞减少性贫血。这种状态不会死亡或者其他并发症;中间型比较少见,外周血象和骨髓象的改变类似重型β地贫;红细胞渗透脆性减低;变性珠蛋白小体阳性;HbA2及HbF含量正常。重型地中海贫血一般在婴儿期几个月时发病,需要依靠终生输血,否则活不过5岁。
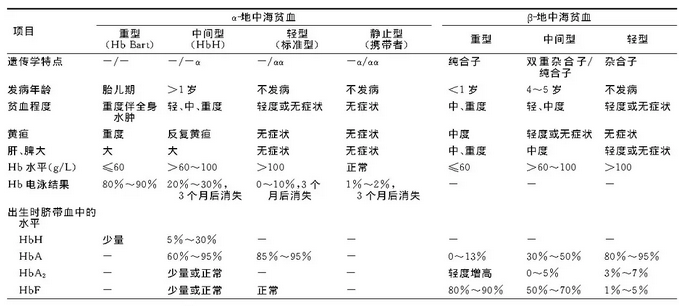
β-地中海贫血的临床特点
β地中海贫血的遗传学变化
β地中海贫血由于基因缺陷造成,是常染色体隐性遗传病。β-蛋白肽链的合成由HBB基因控制,HBB基因位于11号染色体。目前已知的HBB基因达100多种,国内已发现的有28种,常见的突变有:CD41/42(-TCTT )、IVS-2-654(C>T)、CD17(A>T)、-28(A>G)、CD26(G>A)、CD71/72(+A)、CD43(G>T)、-29(A>G)、起始CD(ATG>AGG)、CD14/15(+G)、CD27/28(+C)、-32(C>A)、-30(T>C)、IVS-1-1(G>T)、IVS 1-5(G>C)、CD31(-C)、+40- +43(-AAAC)等。将HBB基因中的一些突变阻止任何β-珠蛋白的产生,即β珠蛋白不存在被称为β-0(B 0)地中海贫血。其他HBB基因突变允许产生一些β-珠蛋白,但数量减少,称为β加(B +)地中海贫血。有B 0或B +地中海贫血并不一定能预测疾病的严重程度。
β地中海贫血的严重程度取决于一个或两个基因受到影响的程度。如果两个基因都受到影响,结果是中度至重度贫血。β地中海贫血的严重形式称为重型地中海贫血。重型地中海贫血患者的父母各自携带一个突变基因的拷贝,但是他们的父母通常不显示病症的体征和症状。然而,有时候,每个细胞只有一个HBB基因突变的人会发生轻度贫血。
这是这种疾病最严重的类型。患病儿童一般在出生后几个月内发病,重型β地中海贫血症可导致严重的并发症,尤其是未经治疗的患者。重型β地中海贫血的并发症包括:
多余的铁:患有β地中海贫血症的孩子可能因为身体内含有过多的铁而死亡,无论是从疾病本身,还是从反复输血。过量的铁会损害心脏,肝脏和内分泌系统。
骨骼畸形和骨折:β地中海贫血可引起骨髓扩张,使骨骼更宽,更薄,更脆。这使得骨骼更容易破裂,并可能导致骨骼结构异常,特别是在面部和头骨的骨骼。
脾脏变大:脾有助于抵抗感染,并从体内滤除不需要的物质,如死亡或受损的血细胞。β地中海贫血可以导致红细胞以更快的速度死亡,使脾脏工作更难,这使得它变大。脾大会使贫血症恶化,如果太大,可能需要排除。
感染:患有β地中海贫血的儿童感染的风险较高,特别是如果他们的脾脏被移除。
增长速度放缓:β地中海贫血导致的贫血可能导致儿童生长缓慢,也可能导致青春期延迟。
重度地贫患着需要频繁的输血,一般3~4周一次,并且可能不能正常的寿命,目前最长的寿命是50多岁。在十几岁的时候,由于频繁输血导致身体中铁过量,过量的心脏和其他器官中,将导致心脏和其他器官衰竭。因此重度地贫患者还需要配合去铁治疗。
1 输血
由于地贫患者的血红蛋白(Hb)合成不足,不能制造有效的红细胞,因而引起慢性血氧不足,代偿性骨髓过度增生,骨髓腔扩张,骨骼变形,肝、脾肿大,脾功能亢进等病变。为维持正常的Hb水平,增进正常生活活动及生长发育,对于重型β-地贫患者应考虑采取长期有规律的输血,目前主张每3~4周输血1次,维持Hb在90~120 g/L之间。对于中间型地贫患者,大多数平时能维持Hb在75 g/L以上,无需依赖长期规则输血,只有在临床症状表现如重型β-地贫时,才需长期规则输血,若感染后,暂时的Hb下降,输血后可回升,对怀孕的中间型患者,怀孕期间需规则输血。
输用洗涤红细胞或去除白细胞后的浓缩红细胞,可减少输血后的过敏反应、肝炎、艾滋病、巨细胞病毒等传染性疾病。在广西,反复输血的小儿丙型肝炎病毒感染率为22.9%。目前临床输血采用ABO血型系统作为配血的主要指标,一般不考虑或不可能考虑HLA配型的问题。反复多次接受含有异己HLA抗原的血制品后,受者体内的HLA抗体会高水平的持续存在。因此在多种先天性疾病的异基因移植中(仅地贫移植前需依赖大量输血),地贫是惟一产生高排斥的疾病。一些临床医生建议给地贫患者输注来自亲属的红细胞以维持生命需要,直到进行异基因移植,使之减少上述风险。
2 铁螯合剂
由于每输注一单位浓缩红细胞(200 ml)将会带入200~250 mg的铁质,因此长期反复输血及骨髓红系细胞造血过盛、肠道吸收铁增加,体内铁负荷过重不可避免,过多的铁沉积于心肌、肝、脑等器官,引起血色病。临床上常出现面色铁青、心力衰竭、肝硬化、生长发育障碍、糖尿病等,其中心脏病变是输血依赖性地贫患者主要的死因。2000年英国有研究报道,50%重型β-地贫患者在35岁以前死亡,主要是因为心脏病变。
应用铁螯合剂进行去铁治疗对地贫患者十分重要,但过早使用铁螯合剂可影响小儿骨骼生长,故常需对体内铁负荷进行评估。最普遍使用的评估方法是测定血清铁蛋白浓度。主张3岁后或接受输血10~20单位红细胞或血清铁蛋白浓度在1000μg/L以上时开始应用去铁治疗。但由于血清铁蛋白易受溶血、炎症、肝病等因素的影响,故数值不够准确;且铁主要沉积在一些高水平表达转铁蛋白受体的组织器官,如肝脏、心脏及内分泌器官,血清铁蛋白仅反映了1%的贮存铁水平;研究显示血清铁蛋白水平与肝脏、心脏铁沉积程度没有很好的相关性。因而仅监测血清铁蛋白、血清铁、转铁蛋白及铁饱和度不足以反映肝脏、心脏等器官的铁沉积情况和去铁疗效。多数学者认为通过活检获得肝铁质浓度是评价体内铁负荷最准确而敏感的指标。但肝穿刺是侵入性检查,具有一定的危险性,在一般的医疗单位,尤其幼年患儿难于进行。目前在欧洲研究者们已将核磁共振检查(T2*)用于检测肝脏铁沉积,结果表明其有效性与肝细胞活检一致。然而并没有较好的技术和资料评定心脏铁沉积情况。
去铁胺(deferoxamine)为六配基铁螯合剂,可和游离的、与铁蛋白和含铁血黄素结合的Fe3+形成铁胺络合物,从而有效地清除肝脏铁沉积,促进铁从尿和粪便排泄。去铁胺是目前使用最广泛而且效果最好的铁螯合剂,也是惟一被批准在美国和加拿大使用的铁螯合剂,已被广泛地使用了20多年。一般予静脉或腹壁皮下缓慢注射,按体重20~40 mg/kg,维持8~24 h,每周5~6 d,用微泵作为动力。若注射时间少于8 h易造成铁螯合剂中毒。临床应用中,去铁胺的不足之处在于:(1)不能进入细胞,仅能清除血液中铁离子;(2)在去除心脏铁沉积、防止心力衰竭和心率失常方面疗效不佳;(3)静脉或皮下注射,维持时间长;治疗费用昂贵。
去铁酮(deferiprone)为二配基铁螯合剂,是最近欧盟批准的口服铁螯合剂,已在43个国家获得批准使用。与去铁胺相比,其优点有:(1)口服螯合剂;(2)可进入细胞,清除细胞内沉积的铁离子;(3)去除心脏铁沉积效果优于去铁胺。去铁酮有效剂量为75 mg·kg-1·d-1,副作用有粒细胞减少及肌肉和关节疼痛、胃肠道反应和锌缺乏。Alan等为评价去铁酮在重型地贫治疗中的有效性和安全性进行了4年的多中心研究。结果显示,中性粒细胞减少的发生率为2.8%,且在无脾肿大的患者中更为明显;对于曾经使用去铁胺治疗后改用去铁酮的患者,血清铁蛋白水平无明显改变;25%的患者因考虑到去铁酮的有效问题而中止治疗。Wu等根据上述两种铁螯合剂的作用特点,联合应用去铁胺和去铁酮治疗重型地贫患者,核磁共振检测表明心脏的铁沉积显著减少,且无明显的不良反应。Tsironi等也报道联合去铁胺和去铁酮使用可以扩大疗效,并减少心脏并发症的发生。目前认为去铁酮是一种口服可短期内有效的铁螯合剂,不如去铁胺安全,长期安全性及效果尚未肯定,在美国未获得批准,仅限用作去铁胺治疗禁忌或重型β-地贫不适合使用去铁胺治疗的铁超负荷的患者。
ICL670是一种新型的口服三配基铁螯合剂的代表,经体内、外模型研究证实ICL670口服后显效,具有良好的耐受性和动员组织铁的功效,可促进铁排泄。Nisbet-Brown等对ICL670的安全性和耐受性、药代动力学及累积铁的清除进行了评估,结果显示:ICL670口服吸收迅速;24 h血液中均可监测到;药代动力学稳定;给予ICL670 20 mg·kg-1·d-1可以清除输注12~15 ml·kg-1·m-1浓缩红细胞所引起的铁沉积,相当于0.3~0.5 mg·kg-1·d-1。Galanello等的研究表明ICL670的血浆半衰期为11~19 h,这支持每日仅需口服用药1次,将有望替代需要每日皮下注射给药的去铁胺。Ⅱ期临床结果显示,ICL670对铁离子具有高的亲合力和选择性,在清除体内过多的铁上和去铁胺一样有效。关于ICL670对成人及儿童输血依赖性贫血患者的铁过载改善作用的Ⅲ期临床研究,正在多个国家同时展开。这是迄今为止对于铁离子螯合剂的规模最大的临床研究项目之一。
3 造血干细胞移植
异基因骨髓移植、外周血干细胞移植及脐血移植是目前根治重型β-地贫的惟一方法。对有HLA相合同胞供者的重型β-地贫患者来说,造血干细胞移植应作为首选治疗。骨髓移植的效果与患者年龄、身体状况、预处理方案、供者来源、HLA相合程度及对并发症的处理等因素密切相关。Lucarelli等根据地贫患者是否肝大,是否肝纤维化,是否规则应用铁螯合剂三种危险因素将患者分为三级:一级为无上述3种危险因素;二级有1~2种危险因素;三级有3种危险因素。最近20年来,由于预防措施的改进、移植相关并发症的有效控制、新的预处理方案的应用,致使HLA相合的同胞移植获得了相当的成功。目前上述三级患者获得HLA配型相合供者的骨髓移植后无病生存率分别为87%、85%和80%。
目前在造血干细胞移植治疗地贫方面备受研究者关注的是造血干细胞的来源问题。从1981年12月到2003年1月共有1003例年龄在1~35岁的地贫患者在意大利Pesaro进行了异基因骨髓移植,地贫患者无病生存大于20年的Kaplan Meier概率为68%。Lawson等统计了英国儿科中心10年(1991-2001年)为55例重型β-地贫患儿(中位年龄6.4岁)进行骨髓移植的情况:移植排斥为4.6%,Ⅱ~Ⅳ度急性移植物抗宿主病(GVHD)发生率为31%,慢性GVHD为14.5%;8年总体生存率和地贫无病生存率分别为94.5%和81.8%。作为造血干细胞来源之一的外周血造血干细胞,因为移植后造血恢复快、移植感染发生率低,且经动员处理后的外周血造血干细胞数目等于甚至超过骨髓,较易获得,现已成为众多研究者选择的对象。但另一方面,由于外周血中包含较多的T淋巴细胞,与骨髓移植相比较,异基因外周血干细胞移植的GVHD发生率较高。Pakakasama等为8例重型β-地贫患儿进行了异基因外周血干细胞移植,移植后中性粒细胞和血小板恢复的中位时间分别为15 d和21 d,4例患儿出现了Ⅱ~Ⅳ度急性GVHD,3例发展成为慢性GVHD。所有的患儿随访23(7~52)个月仍存活,没有出现移植失败或移植物排斥。Yesilipek等观察15例进行异基因外周血干细胞移植的患者,中性粒细胞和血小板恢复的中位时间为12 d和16 d,3例出现慢性GVHD,其中2例死亡,随访13例患者2~30个月,病情稳定无地贫复发,总体存活率和无病生存率为86.6%。考虑到异基因骨髓移植的相关死亡率和慢性GVHD发生率较高。中山大学第二附属医院儿科完成的9例脐血移植治疗重型地贫病例,经过49(38~64)个月的随访,4例治愈,4例复发,仍需输血。只有不到30%的患者可以从亲属中获得HLA配型相合的造血干细胞,因而无关供者的造血干细胞成为一部分研究者的选择。国际骨髓捐献目录中登记的4百万志愿捐献者可以为患者提供无关供者造血干细胞。曾在该目录中为272例地贫患者寻找可能匹配的供者,结果显示80.2%的患者找到了至少1个6/6 HLA匹配的供者或脐血,所有的患者都找到了5/6 HLA匹配的供者或脐血,这使无关供者造血干细胞移植治疗重型β-地贫成为可能。Hall等报道使用4/6 HLA匹配的无关脐血成功治愈1例2个月大的重型β-地贫婴儿,目前该患者为移植后5年,生长发育正常,为供者染色体。国内中山大学第二附属医院儿科为1例5岁的重型β-地贫患儿进行了6/6 HLA匹配的无关脐血移植,移植后出现轻度肝静脉闭塞症、急性Ⅲ度GVHD、巨细胞病毒间质性肺炎等并发症,经相应治疗后病情得到控制,随访20多个月仍为治愈状态。
为解决造血干细胞来源的问题,一种新型的遗传诊断技术——胚胎植入前HLA配型逐渐发展起来。该技术可为地贫患者挑选正常的、HLA配型相合的婴儿供者。美国研究者首次在临床中证明此项技术具有可行性,因孩子患白血病或贫血性疾病的9对夫妇参与了这项研究。研究者通过体外授精培育199个胚胎,HLA检测发现,其中45个胚胎分别与患者匹配,这些匹配胚胎中28个随后被植入子宫,最终有5名健康婴儿顺利出生,其中1名婴儿捐献的脐血干细胞已被用于治疗其患病的同胞。这项技术可以确保新出生婴儿捐献的干细胞能够与患病同胞相匹配,移植后不会在患者体内遭到排异。除白血病和贫血性疾病外,该技术还可能用于治疗其他一些恶性疾病,给那些无法为自己患病孩子找到理想干细胞捐献者的父母提供帮助。2004年Cyprus国际会议上介绍了胚胎植入前HLA配型在造血干细胞移植方面的应用情况,及相关的经验。目前通过该技术已使25名妇女怀孕,顺利出生了14名与其同胞HLA匹配的婴儿。
4 药物治疗
β-地贫患者的常规治疗手段依赖于定期输血及使用铁螯合剂,但昂贵的费用对于大多数发展中国家的患者是沉重的负担。为此,通过药物刺激或重新激活γ-珠蛋白基因的表达,增加胎儿血红蛋白(HbF)的合成,改善α与非α珠蛋白链的合成失衡已成为目前临床上治疗β-地贫和Hb病的可行手段。有关药物诱导γ-珠蛋白基因激活的机制主要有两大类:一类是通过红系细胞分化动力学改变来增加γ-珠蛋白基因表达,如红系刺激因子,以及以羟基脲为主要代表的一系列细胞毒类药物;另一类是直接激活γ-珠蛋白基因启动子或其他可能增强的位点,使γ-珠蛋白基因激活,包括组蛋白去乙酰化酶抑制剂和去甲基化剂。
羟基脲(hydraxyurea)是细胞周期S期特异性抑制二磷酸核糖核苷酸还原酶的细胞毒性抗肿瘤药物,临床上已广泛用于治疗慢性髓系白血病及其他骨髓增殖性疾病,是第一个通过美国FDA批准用于镰状细胞贫血治疗的药物。羟基脲在激活γ-珠蛋白基因表达方面具体的作用机制尚未明确。
丁酸及其衍生物(butyrates)是新一类组蛋白去乙酰化酶抑制剂。在治疗地贫方面,丁酸盐类可明显抑制Hb的转换过程,提高非α链/α链比值,刺激HbF合成,诱导γ-基因活化作用呈剂量依赖性反应。丁酸及其衍生物是短链脂肪酸类化合物的代表药物,主要包括丁酸钠、正丁酸、α-氨基丁酸、苯丁酸钠、异丁酰胺等。该类药物具有低毒性,但由于其极短的半衰期等因素限制了临床应用。
5-氮胞苷(azacytidine)是一类甲基转移酶抑制剂,可引起DNA的去甲基化。5-氮胞苷是最先用于临床通过活体内激活γ-珠蛋白基因的策略治疗β-地贫的药物。治疗效果显示患者骨髓组织中γ-mRNA转录被激活,纠正了珠蛋白链的不平衡,患者甚至可以不再需要输血。然而5-氮胞苷具有较强的毒性作用,主要是骨髓抑制和中性粒细胞减少,并有潜在的致癌作用,因此从20世纪80年代中期有关5-氮胞苷的研究已基本停止。
5 脾切除及脾动脉栓塞
对巨脾或及脾功能亢进者可行脾切除术或脾动脉栓塞术,以减轻溶血。切脾指征:(1)脾大6 cm以上或脾功能亢进;(2)每年输血量超过200~250 ml/kg红细胞者;(3)5岁以上(5岁以前小儿机体免疫功能发育未完善,术后常并发严重感染)。脾切后患者输血量减少,红细胞寿命延长,贫血症状改善。目前认为HbH病、中间型β-地贫、β-地贫复合HbE病患者脾切效果好,脾切后因免疫功能减低容易合并感染,同时血小板明显增高,易导致血栓栓塞,肝脏含铁血红素沉积加重并明显增大,其他器官亦受累。脾切后应立即给予抗生素预防感染1~2个月。血小板>800×109/L者应给予阿司匹林、潘生丁等抗凝治疗。
6 基因治疗
随着对地贫发病机制的深入研究,地贫成为了最早被尝试用于基因治疗的疾病之一,并且近20年来在地贫的基因治疗方面取得了很大进展。一般基因治疗的模式是将地贫患者的造血干细胞取出,在体外导入并整合正常的珠蛋白基因后再植入患者体内,使转移的珠蛋白基因随干细胞分化获得红细胞特异的高水平表达,达到治疗地贫的目的。研究证明,应用慢病毒载体(lentivirus vectors)可将活化的内源性位点调控区元件与β-珠蛋白基因共同导入造血干细胞,在动物模型实验中得到β-珠蛋白基因的高效表达。2000年May等首次报道将带有大片段调节序列的人β-珠蛋白基因的慢病毒载体转染鼠骨髓细胞,人的β-珠蛋白基因准确而有效地整合到宿主DNA中,高效表达,合成的正常Hb达到治疗β-地贫鼠的目的。最近,Puthenveetil等采用慢病毒载体,将β-珠蛋白基因转导入人的重型地贫CD34+细胞,并在细胞培养中得到了一致的红系分化,纠正了异常的地贫特征,然后将培养的细胞移植入免疫缺陷鼠,3~4个月后仍有正常的红系表达。
7 预防
重型β-地贫目前总体预后很差,多数于学龄前因继发感染、全身及心力衰竭而死亡。总体上临床控制的效果仍不理想。因此,预防方面的控制显得尤为重要。预防控制的主要措施包括通过社区筛查、遗传咨询和产前诊断手段控制重型β-地贫患儿的出生。
地贫的防治工作是一项系统工程,需要全社会的支持和干预,实施地贫预防与监护计划,积极开展社会宣教、人群筛查和重点筛查、遗传咨询、产前诊断等工作。只有通过这些努力,才能真正实现降低地贫的发生率,提高人口素质,减轻社会和家庭负担并保障孕妇的身心健康的目标。
治疗进展
2.1 非药物治疗-异基因造血干细胞移植(Allo-HSCT)
Allo-HSCT仍然是目前唯一有可能明确治愈该疾病的非药物治疗。并作为重型地中海贫血患者的基因替代疗法已经得到了很好的确立, 现在为接受这种治疗的患者提供了非常高的治愈率。骨髓是干细胞的优选来源, 正在尝试使用外周血干细胞以降低高风险患者移植排斥的风险.而事实上, 骨髓和干细胞移植由于配型和髓源限制、实施中的风险以及昂贵的费用, 很难在临床上普及。
2.2 药物治疗
2.2.1 改善红细胞无效生成
有研究者观察到β-地中海贫血的患者骨髓中含有多于健康对照者5-6倍的红系前体的数目, 嗜碱性和多色性成红细胞增加, 正染色性成红细胞减少。这结果说明在β-地中海贫血红细胞生成过程中, 早期阶段的红细胞前体(成红细胞原) 的不断扩张, 加上骨髓中红系分化加速和成熟障碍, 故而导致红细胞前体细胞凋亡率显着增加, 引起了无效的红细胞生成。因此, β-地中海贫血患者外周血中地中海贫血红细胞的输送量大大降低。
Jak2抑制剂和TGF-β超家族均可降低输血依赖性β-地中海贫血患者输血次数, 减少治疗成本和输血过多引起的铁过载风险, 进一步提高患者生活质量。
2.2.1. 2 JAK2抑制剂
在β-地中海贫血中, 无效的红细胞生成导致贫血, 缺氧, 并通过JAK2/STAT5途径增加EPO(促红细胞生成素) 产生。促红细胞生成素水平升高导致携带磷酸化Jak2的红细胞前体的数量增加。这被认为是无效的红细胞生成, 伴有大量红细胞增生和髓外造血。因此, JAK2抑制剂可以缓解非输血依赖性地中海贫血的脾肿大和无效红细胞生成的情况。β-TI(β地中海贫血中间型) 小鼠模型的研究表明, JAK2抑制剂的短期给药降低了红细胞生成和脾肿大的发生率。鲁索替尼是JAK2抑制剂, 可降低STAT(信号转导子和转录激活因子) 家族蛋白, 特别是STAT5和STAT3的磷酸化。STAT5的磷酸化能促进基础红血细胞生成, 并在应激红细胞生成期间使其加速, 提高血红蛋白水平。目前干预定期输血的地中海贫血患者的2期临床试验中正在进行。
其安全性和耐受性研究正在进行, 短期可能改善仅靠输血缓解贫血的现状, 可能缩短治疗时间, 有助于提高患者生活质量。
2.2.1. 3 转化生长因子-β(TGF-β) 超家族
TGF-β超家族由四组在细胞水平上具有相似结构和调节活性的蛋白质组成:TGF-β, BMP(骨形态发生蛋白) , GDFs(生长和分化因子) 和激活素。TGF-β超家族在调节诸如发育, 分化和组织稳态等基本生物过程(包括骨和造血组织) 中发挥重要作用。GDFs或激活素的抑制剂可能对红细胞生成的晚期具有积极的影响, 在由无效的红细胞生成引起的贫血, 如β-地中海贫血等疾病中具有治疗作用。来自激活素受体(ActRIIA或ActRIIB) 的修饰受体, 其细胞外部分可与IgG免疫球蛋白的Fc部分的融合, 起到抑制Smad2/3通路传导的作用。相对于激活素或其它TGF-β家族成员(例如GDF11) 来说, 它们具有更强的结合能力。目前正在进行的有关激活素受体(ActRIIA或ActRIIB) 的药物Sotatercept和LuspaterceptⅠ/Ⅱ的临床研究结果显示血红蛋白水平稳定增加, 贫血得到剂量依赖性的改善, 且药物安全性较好, 且治疗费用较低。Sotatercept和Luspatercept目前仍有不足:其最佳治疗剂量和剂型尚未确定。针对β-地中海贫血无效红细胞生成的3期临床试验正在进行, 有望解决这一不足。
这两个药的出现一方面丰富了因无效的红细胞生成导致贫血的β-地中海贫血治疗手段, 根据患者不同的贫血情况选用不同的方案。另一方面有望成为新的可选择的治疗手段。
2.2.2 铁螯合治疗
铁超载(IO) 是地中海贫血患者的一个主要问题, 铁过载在地中海贫血的主要原因是反复输血, 影响包括肝脏, 心脏和心脏在内等实质器官。铁稳态主要由肝脏中产生并由HAMP(铁调素抗菌肽) 基因编码的蛋白质hepcidin(铁调素) 严格调控。肠的刷状缘细胞, 网状内皮系统的巨噬细胞和肝细胞是铁调素的靶细胞。铁调素降解铁转运蛋白, 用于将铁从细胞内转运至细胞外空间, 抑制了肠道中铁的吸收和巨噬细胞和肝细胞中储存的铁的释放。同时铁调素还可以改善无效的红细胞生成。Hepcidin对细胞内和细胞外铁浓度的调控发挥了重要作用。铁调素产生受到各种不同的因素影响包括红细胞生成增加, EPO水平升高, 炎症反应等。有临床观察示去铁胺联合去铁酮治疗重型β地中海贫铁过载, 效果显著, 且安全、无严重不良反应。虽然不同种类去铁剂联合应用可有效降低血清铁蛋白水平, 改善脏器功能, 但除增加患者经济负担外, 还会影响患者的治疗依从性。因此, 研发治疗简便、副反应少的新型去铁剂是必要的。
2.2.2. 1 转铁蛋白
试验发现转铁蛋白注入β地中海贫血小鼠可使红细胞存活率正常化, 循环血红蛋白浓度增加, 网状红细胞增多较前减少, 血清促红细胞生成素水平降低, 可逆转脾肿大, 循环游离血浆铁水平恢复正常, hepcidin表达增加。且全部小鼠在注射过程中幸存, 没有明显的不良影响。转铁蛋白可从促红细胞生成机制出发, 利用贮存铁和实质沉积铁来合成血红蛋白, 从而消除无效的红细胞生成。
目前有关转铁蛋白的提取制作并未公布, 相关基础研究和疗效观察较少。可以进行样本量较大的动物实验或小样本量的临床观察确定其安全性和耐受性。注入转铁蛋白可以加快铁蛋白的代谢, 有效地减少因铁过载引起的并发症。其与药物相比, 不良反应估计较后者少, 不过这个有待证明。
2.2.2. 2 抗氧化治疗
体内过量的铁会产生自由基, 从而引起氧化应激, 导致细胞损伤, 甚至细胞死亡。有研究认为氧化应激(氧化损伤) 是地中海贫血中细胞损伤和组织损伤的主要原因之一。在地中海贫血患者中, 贫血引起氧合血红蛋白不稳定, 从而造成红细胞膜上产生内部活性氧(ROS) 。位于质膜上的ROS因为不易被细胞质抗氧化系统接触, 所以可以容易地氧化膜脂和蛋白质, 对细胞及组织造成广泛的氧化损伤, 并破坏磷脂的生理学对称性, 暴露磷脂酰丝氨酸。而磷脂酰丝氨酸具有血小板功能因子-3(PF-3) 样活性, 被认为是血小板激活剂。磷脂酰丝氨酸的外化和脱落由增加的细胞Ca2+介导, 并对红细胞衰老起重要作用。磷脂酰丝氨酸会促进血管炎症部位的血栓形成, 引起血管阻塞, 组织缺氧, 甚至死亡。已经证实β地中海贫血患者红细胞磷脂酰丝氨酸暴露增加与红细胞自我衰亡相关。
白藜芦醇作为一种多酚, 用于治疗β-地中海贫血症。鉴于其促进末端红细胞分化与FoxO3a活化的新机制, 利用其作用和抗氧化特性, 通过过氧化氢酶和Prdx2, 起到降低β-地中海贫血无效红细胞生成的作用。
水飞蓟素是从乳蓟中分离的黄酮木脂素复合物, 属于植物类的酚类化合物, 具有抗氧化和清除自由基的能力, 并显示出对铁离子呈现出强烈的亲和力。研究显示水飞蓟素可以用于抵抗由于氧化应激引起的自由基损伤。且具有免疫调节作用, 并呈剂量依赖性。
据研究, 脂质抗氧化剂(维生素E) 与蛋白质抗氧化剂(N-乙酰基半胱氨酸) 和铁螯合剂(去铁酮) 的组合使用比用单一抗氧化剂有效, 可减轻患者氧化应激, 减少氧化应激, 促进血红蛋白浓度升高, 改善高凝状态。
抗氧化治疗可以减少细胞及组织损伤, 减少并发症的产生。在缺少铁螯合剂情况, 抗氧化剂可作为铁螯合剂治疗, 未来可以作为地贫铁螯合治疗的新方案。
2.2.3 调节珠蛋白失衡
2.2.3. 1 表观遗传学基因修饰治疗
目前这种治疗中只有增加β球蛋白的项目在地中海贫血患者中得到试验。输血依赖性患者已经尝试β-珠蛋白载体, 体外转导自体的CD34+红细胞祖细胞移植, 其安全性和耐受性也在试验中进行评估。β-地中海贫血多数患者的β-珠蛋白表达降低, 与之相对应的α-珠蛋白链过量介导积累, 容易损伤红细胞及其前体。许多病例对照研究和队列研究已经证实, β地中海贫血出现的同时, α珠蛋白链输出的也会降低。近30年来报道的临床遗传学数据越来越多地证明α-珠蛋白表达降低25%-50%, 可以改善β-地中海贫血患者的临床严重程度。显然通过RNA干扰, 表观遗传药物靶向或基因组修饰, 治疗性地下调α-珠蛋白表达是可行的。有研究确定泛组蛋白脱甲基酶抑制剂IOX1, 其能选择性下调α-珠蛋白表达而不干扰β珠蛋白表达。
2.2.3. 2 胎儿血红蛋白(HbF) 诱导剂
有研究者提出β-地中海贫血的一种非常有希望的治疗方法是胎儿血红蛋白(HbF) 通过药物重新激活内源性γ-珠蛋白基因产生, 可以作为γ-珠蛋白来替代缺失或减少的成人β-珠蛋白。因为HbF诱导剂通过与过量的α珠蛋白链结合产生胎儿血红蛋白(HbF) 来降低潜在链的不平衡。
羟基脲(HU) 是最广泛接受的HbF诱导剂, 其S相特异性和非DNA-低甲基化化疗剂能够诱导HbF合成。羟基脲用于输血依赖性β地中海贫血患者中, 可以减少患者的输血次数的同时, 还可以有效地提高血红蛋白水平。HU耐受性良好, 无长期的不良反应, 仅伴有轻度和短暂的不良事件。有研究表明羟基脲联合小剂量维生素C治疗中间型β地中海贫血, 疗效显著, 患者无明显不良反应。
地西他滨(5-氮杂-2’-脱氧胞苷) 显示去甲基化并可以重新激活g-珠蛋白基因的表达。在低浓度下, 它不会造成显著的DNA损伤或细胞毒性。2011年的一项初步研究显示, 5例中间型地中海贫血患者皮下注射地西他滨0.2 mg/kg, 每周2次, 共12周, 患者贫血情况得到改善。但超低剂量地西他滨联合羟基脲治疗中间型β地中海贫血效果欠佳, 对改善血红蛋白水平有限, 但可明显升高血小板。
鲁索替尼作为生成HbF的诱导剂, 也是JAK2抑制剂, 但其STAT3可抑制γ-珠蛋白基因的表达。STAT3磷酸化的减少可以减少γ-珠蛋白基因表达的抑制。为此, 可用于治疗血红蛋白病, 特别是在对HU治疗无应答或在长期治疗后表现出低应答的患者。
口服给药的丁酸盐化合物(苯丁酸钠和异丁酰胺) 对少数β-地中海贫血患者以及儿童可有显著的刺激HbF合成作用, 具体诱导的机制尚未完全阐明。由于丁酸盐是组蛋白脱乙酰酶的抑制剂, 因此提出丁酸盐可通过组蛋白乙酰化的改变, 来提高γ-珠蛋白基因的转录速率, 进而提高HbF水平。丁酸盐可以通过增加γ-珠蛋白的翻译效率来诱导HbF mRNA的表达。
β-地中海贫血多是因为β-珠蛋白缺失, HbF生成诱导剂可以在病源上通过消耗过量的α珠蛋白链来降低潜在链的不平衡。目前研究均证明临床有效, 且较少不良反应, 但未有进展性研究。HbF诱导剂可以在β-地中海贫血未出现贫血等症状前干预治疗, 有利于减少住院率, 提高患者生活质量。
β-地中海贫血的基因治疗
1 β-地中海贫血的药物基因调控治疗
在正常情况下, 由胎儿到成人珠蛋白合成的发育开关于出生后12周内即已完成。随着γ-珠蛋白基因表达的逐渐关闭, 重型β-地中海贫血的患者开始出现临床症状。药物基因的调控治疗是用药物来调控珠蛋白基因的表达, 合成γ-珠蛋白肽链以提高人胎儿血红蛋白(human fetal hemoglobin, HbF) 。临床上很早就注意到, β-珠蛋白肽链分子结构或者合成具有严重遗传缺陷的患者, 如果能产生高水平的HbF, 则常常只有很缓和的贫血症状。因此, 提高HbF可以有效治疗地中海贫血。迄今为止, 所有已经得到的基础实验研究及临床评价的试剂, 可以通过两种途径实现在成人期重新活化HbF的合成。①通过调节成人红系干细胞分化过程中的“压缩性开关”影响F细胞的生成, 主要是改变红系增殖的动力学状态并“募集”保留γ-珠蛋白链合成程序的红系干细胞群进入增殖和分化, 这种红系干细胞群在红骨髓中保持静止状态, 但在急性红系增生的刺激下进入早熟分化。代表此类的药物包括EPO等造血生长因子, 以及以羟基脲为代表的一系列细胞静止剂。②直接作用于γ-珠蛋白基因启动子或其他可能的正性作用调控元件以活化γ-珠蛋白基因的表达。通过第二条途径再活化γ-珠蛋白基因的典型代表是丁酸盐及其衍生物。
1.1 5-氮胞苷
使用药物手段在活体内操作并增强胎儿珠蛋白的表达, 首先是在用5-氮胞苷刺激贫血狒狒合成HbF的工作中得以证明的。为数不多接受5-氮胞苷治疗的患者几乎全部显示了明显的效果, 骨髓组织中的γ-mRNA转录被激活, 珠蛋白链的不平衡被纠正, 以及血红蛋白水平的显著增高, 长期接受5-氮胞苷治疗的患者甚至可以不用输血。5-氮胞苷治疗地中海贫血的机制最初被认为是低甲基化的状态, 在后来的研究中发现, 在离体培养于5-氮胞苷中的红系干细胞在重新铺板于不含药物的培养基12h后可以继续表达HbF, 显示上述重编分化程序的模型可能产生作用, 表明5-氮胞苷的作用是多因素的。然而, 5-氮胞苷具有较强的毒性, 主要是骨髓抑制和粒细胞减少, 常并发感染。作为化疗药物, 5-氮胞苷自身具有很高的诱变性, 具有潜在的致癌作用。在20世纪的80年代基本研究停止。
1.2 羟基脲
羟基脲是抑制二磷酸核糖核苷还原酶S期特异性细胞毒药物, 临床上已广泛用于治疗慢性髓系白血病及其他骨髓增殖性疾病, 是第一个通过美国食品药品监督管理局批准用于镰状细胞贫血治疗的药物。由于镰形细胞贫血和β-地中海贫血在发病机制上存在着相似性, 人们也尝试将其用于β-地中海贫血的治疗。在对灵长类实验动物和镰状细胞贫血的患者进行的试验中显示了羟基脲诱导HbF合成的显著能力, 并明显改善了患者的临床症状, 但是, 在治疗地中海贫血方面疗效并不确切, Watanapokasin等报道了羟基脲治疗的18例β-地中海贫血和血红蛋白患者, 治疗后检测其γmRNA的水平, γ/αmRNA平均水平增加了9倍。其中4例患者的HbF增加了30%, 9例患者的HbF增加了20%~30%, 剩下的5例患者的HbF增长<20%, 其中的2例<3%。可见对于羟基脲治疗β-地中海贫血, 不同患者的敏感程度不同。羟基脲诱导HbF合成的最大效能是在产生了骨髓抑制的毒性作用情况下实现的, 表明存在对F细胞的选择作用。但是羟基脲不能在离体培养的晚期干细胞中诱导胎儿γ-珠蛋白的表达, 所以没有观察到重新编制红细胞分化程序的作用。因此羟基脲对珠蛋白基因的表达的作用可能比以前所认为的要广泛些, 其缺乏对胎儿珠蛋白基因的特异性诱导使之难以纠正β-地中海贫血的珠蛋白肽链的不平衡。而且使用羟基脲的若干问题并未解决, 长期使用该药物对人体的毒性仍然令人担忧, 包括患者对药物的清除能力, 药物应答的可预见性, 以及相关器官被羟基脲损坏的可逆转性等。
1.3 丁酸盐类药物
丁酸盐是一种4-碳的短链脂肪酸, 主要有丁酸盐、苯丁酸盐、丙酸盐等几类, 因为其具有较高的安全性, 近来被应用于地中海贫血的治疗, 其根本机制也是促进HbF的生成, 代偿β-珠蛋白肽链生成不足导致一系列红细胞的损伤。早在1975年在丁酸钠处理小鼠红白血病细胞中就观察到的联苯胺染色反应阳性, 表明丁酸盐能够诱导血红蛋白合成。此后, 对实验动物模型、正常人(包括新生儿) 以及β-地中海贫血和镰型细胞贫血的患者(包括新生儿) 进行的红系细胞离体培养及体内诱导试验证明, 丁酸及其衍生物选择性刺激胚胎或胎儿珠蛋白基因, 并且这种作用并不仅限于丁酸盐衍生物, 而是短链脂肪酸类化合物和其他丁酸盐结构类似物的共同性质。RNA分析表明, 丁酸及其衍生物或短链脂肪酸化合物诱导珠蛋白基因表达的作用一般表现在转录水平上。对于γ-珠蛋白基因, 尚未发现能够在启动子内确定短链脂肪酸类化合物反应模体。在丁酸盐触发的信号传导途径上游也显示涉及几种可能途径。作用机制并不完全明确。Perrine等首先进行的实验性治疗报道包括全部的6例重型β-地中海贫血以及镰状细胞贫血的患者的γmRNA水平都有显著上升(2~6倍) , 与处理前水平相比γ/γ+β珠蛋白合成比例也有6%~45%的增长, F细胞高于基线水平的1.5~3倍。在对一批镰状红细胞贫血患者的为期6个月的治疗中, F网织红细胞和HbF浓度均有迅速增加。然而, 对11例纯合子型β-地中海贫血的治疗中尽管少数患者血红蛋白有少数增加, 但其α/非α-mRNA比例无明显改变, 而且HbF的绝对浓度水平亦无变化。
总的来说, 除了长期输液和服用不便等使用上的问题外, 丁酸盐类药物的毒副作用很小。与上述药物情况相似, 迄今使用的丁酸盐类药物的临床使用也并未能取得稳定一致的效果。同种药物的诱导作用不能在多数患者中得以重复以得到定论, 各方面的研究都需要进一步的探索。
2 β-地中海贫血的基因矫正治疗
地中海贫血发生的根本原因是β-珠蛋白基因及其调控序列由于突变而导致珠蛋白合成减少, 结果是原来在数量上与之持平的α-珠蛋白肽链相对过剩, 在红细胞聚集成包含体, 导致未成熟的红细胞或者成熟的红细胞裂解而出现贫血症状。针对β-地中海贫血发生的分子机制, 许多实验室采取的基因治疗策略是向患者的造血干细胞中导入正常的β-珠蛋白基因补充β-珠蛋白表达的不足或替换异常的β-珠蛋白基因来达到治疗的目的。
2.1 补偿策略的珠蛋白基因治疗
β-珠蛋白基因向造血干细胞的高效转移以及高效表达是β-地中海贫血基因治疗的核心问题。
以逆转录病毒为载体转移目的基因时, 用目的基因取代病毒结构基因而保留ψ包装序列及LTR序列再转染包装细胞, 后者含所有病毒结构基因而缺失ψ包装序列, 在功能上正与逆转录病毒载体互补, 因而可以将逆转录病毒载体包装成具有一次性感染的病毒粒子, 可用于感染靶细胞。
腺病毒相关病毒载体由细小的DNA病毒改造而成。与逆转录病毒载体相比, 具有以下特点:可以转染分裂细胞又可以转染非分裂细胞, 在宿主体内以定向整合的方式存在, 70%以上的整合位点位于第19号染色体q13.3~qter区, 且对人体无致病性, 故AAV重组体在细胞内能长期稳定地表达, 还可避免随机整合可能引起的抑癌基因失活和原癌基因激活的危险, 且在体内不引起明显的病理变化, 表明AAV是一种很有前途的基因治疗载体。
慢病毒属逆转录病毒科, 为二倍体RNA病毒, 分为灵长类病毒如人类免疫缺陷病毒1、猴免疫缺陷病毒和非灵长类病毒如马传染性贫血病毒, 但它与逆转录病毒不同, 能感染非分裂细胞。目前研究较多的是来源于人类免疫缺陷病毒1的慢病毒载体。HIV载体具有能感染非分裂期细胞、容纳外源性目的基因片段大、基因持续表达、免疫反应小等特点。大量研究已表明, HIV载体可以较容易的感染一些用其他载体较难进行转基因的组织, 并且不会引发明显的免疫反应。
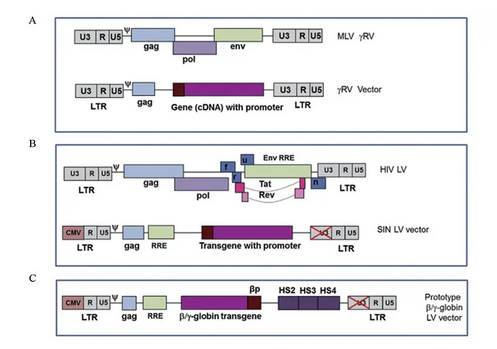
近些年来研究表明, 位于β-珠蛋白基因簇ε-珠蛋白基因基因上游6~22kb的位点控制区(long control region, LCR) 是β-珠蛋白基因簇各功能基因必不可少的远端调控元件。LCR由4个红系组织特异的和发育阶段稳定的DNaseⅠ高敏位点(5′HS1~HS4) 组成。每个HS均由进化上保守的核心序列和侧翼序列组成。转基因动物和细胞转染实验证实完整LCR使与之相连的珠蛋白基因获得红系特异的拷贝数依赖性和整合位点非依赖性的高水平表达。总之, 在β-珠蛋白基因转移的载体中, 不但需要完整的结构基因和近端调控程序, 还必须引入LCR才能提高转移的β-珠蛋白基因在红系细胞中的表达水平。
但是目前, 所有的病毒载体系统虽有较高的转移效率但载体容量有限(8kb和5kb) , 而完整的β-珠蛋白基因及表达需要的近端调控顺序长约3kb, β-珠蛋白基因表达所需的LCR长度达16kb, 远超出病毒载体的容量极限。
各实验室在构建病毒载体的时候都利用能部分代替LCR活性的单一HS或不同活性的HS组合, 称为miniLCR。Sadelain等用长度介于200~400bp的HS2~HS4核心序列构建了约1kb的miniLCR, 插入β-珠蛋白逆转录病毒载体能稳定整合于红系细胞并表达平均水平与内源α-珠蛋白可比的β-珠蛋白, 但各细胞克隆的β-珠蛋白水平呈现较大差异, 说明1kb LCR不能完全克服位置效应的影响使β-珠蛋白基因获得完全的表达。该重组在长期造血移植小鼠造血组织表达了5%~20%的β-珠蛋白。May等首次报道, 将携带珠蛋白基因和调控序列插入慢病毒载体中, 转染到鼠的骨髓细胞中, 高效的表达珠蛋白基因, 纠正了β-地中海贫血鼠的地中海贫血表现。Puthenveetil等把长度为3.1kb的LCR片段和2.3kb的珠蛋白基因片段插入慢病毒载体, 该重组体在造血干细胞移植的β-地中海贫血鼠中得以高效表达, 血红蛋白水平从(2~3) ×103g/L稳定增加到(10~11) ×103g/L。达到了治疗β-地中海贫血鼠的作用。慢病毒载体携带珠蛋白基因和LCR片段时, 由于携带的序列较长, 转染的稳定性并不肯定。Arumugam等设计慢病毒载体时插入了cHS4, 相当于隔离子的作用, 与原来没有隔离的载体相比, 人珠蛋白基因的表达增加了2倍, 减少了位置效应, 增加了载体的稳定性。
多数实验室利用β或γ基因作为病毒的结构基因, 来替代缺陷的珠蛋白基因, 目前大部分研究工作依然处于优化载体的构建, 提高转染的效率和载体的稳定的阶段。
2.2 取代策略的基因治疗
用于校正缺陷的珠蛋白基因以能解决其低水平表达问题的理想方法是利用同源重组技术进行基因打靶, 用正常的顺序代替不正常的DNA区。一个突变的β-珠蛋白基因(βS) 已经在细胞系中实现了体外校正, 然而, 这一方法目前还不能用于骨髓干细胞, 因为正确的位置的重组频率是很低的, 并且需要对过于稀少的被矫正的干细胞进行选择和扩增。
2.3 反义策略的基因治疗
反义RNA是能抑制基因表达的RNA, 它本身含有一段与靶基因互补的序列, 能与靶基因所编码的mRNA结合, 形成反义RNA-靶RNA聚合体, 从而影响靶RNA的功能。其一, 反义核酸技术通过对突变mRNA前体剪接加工的修饰作用, 达到恢复正常剪接的目的。Gong等构建特异性针对βIVS-2-654 C→T异常剪切位点的反义核酸表达载体, 转染培养的红系细胞。特异性的反义核酸能够纠正红系细胞βIVS-2-654 C→T异常剪切模式, 恢复正常的剪接途径, 改善珠蛋白合成的不平衡。其二, 通过与目标DNA双链结合, 阻断特定基因的复制与转录, 或者与目标mRNA结合, 这种结合既可以激活细胞内切酶降解mRNA, 也可阻碍mRNA的剪切, 翻译, 通过上述机制, 纠正异常基因的表达, 调整珠蛋白合成比例的不平衡。
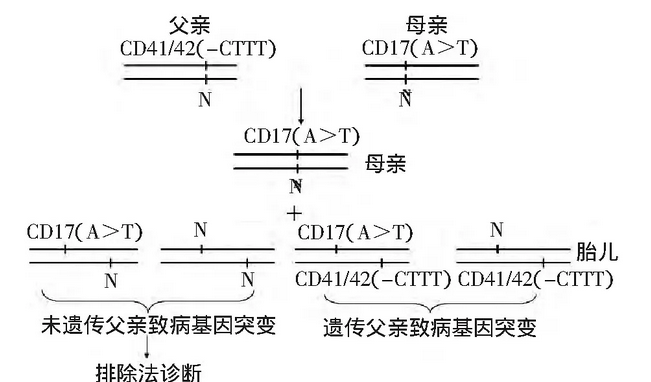
双亲携带异型突变的排除法诊断策略
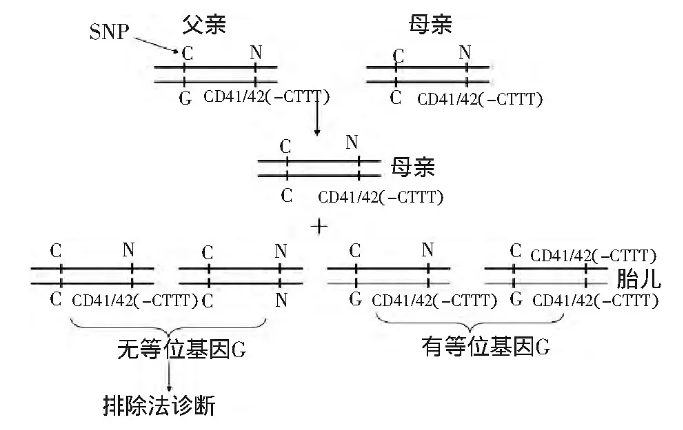
双亲携带同型突变的排除法诊断策略
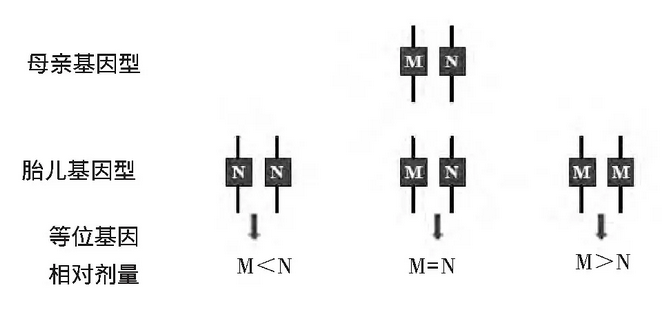
双亲携带同型突变的单倍型相对定量诊断策略
----
αβ复合型地中海贫血
临床上α和β地中海贫血是最主要的地中海贫血类型, 据现有流行病学调查资料显示, 我国广东、广西、海南、台湾和香港等地区人群中α地中海贫血发生率高达4%~15%;β地中海贫血约为1%~6%。相同类型地中海贫血杂合子间的婚配, 生育重型地中海贫血患儿的几率为1/4, 不同类型地中海贫血(α与β) 杂合子婚配, 没有生育重型地中海贫血儿的风险, 但生育αβ复合型地中海贫血双重杂合子的几率为1/4。这种双重杂合子虽本身没有严重的临床表型, 但无论与α或β杂合子婚配, 均有可能生育重型地中海贫血儿, 即胎儿水肿综合征或中、重型β地中海贫血儿, 风险率均为1/4。在我国, α地中海贫血基因的携带率高于β地中海贫血基因的携带率, 广东地区和广西地区α地中海贫血携带率分别为8.53%和14.95%。流行病学调查显示, 广东省人群中同时携带α和β地中海贫血基因突变的比例为0.26%。因此, 提高αβ复合型地中海贫血检出率, 对指导地中海贫血遗传咨询和准确进行产前诊断都具有重要意义。
本组资料显示, 轻型复合型患者有小细胞低色素表现, 但其贫血并不严重, 为轻度贫血或正常, 与单纯的β地中海贫血或α地中海贫血携带者差别不大。此类患者的血红蛋白电泳结果与单纯的β地中海贫血携带者无异, 受α地中海贫血影响较小, 轻型复合型患者的表型和β地中海贫血杂合子一样, 即使复合了α地中海贫血, 也没有受到影响。我们认为这一现象与Hb的肽链组成有关。成人体内Hb有以下3种:HbA(α2β2) , HbF(α2γ2) 和HbA2(α2δ2) , 每种Hb都由2个α珠蛋白肽链和2个非α珠蛋白肽链组成。正常成人个体HbF含量<1%, HbA2含量在2.5%~3.5%, HbA含量最高。轻型复合型患者的α肽链和β肽链合成均降低, 但因HbA占比例高, 其合成降低的幅度也就大大超过HbA2, 因而此类患者表现为HbA2的比例相对增高, 即类似于轻型β地中海贫血。
我们同时发现1例β地中海贫血杂合子复合HbH病的患者, 其表现为轻度贫血, 但血红蛋白电泳结果中出现1.4%HbCS, 1.6%Hb Bart's, 伴HbA2减低为1.6%, 这可能是因为α地中海贫血基因中和β地中海贫血表现的程度依赖于失活的α珠蛋白基因个数。通常失活1个α基因对β地中海贫血表现没有明显影响, 而失活3个(HbH病) α基因对β地中海贫血的临床表现影响就会很明显, 因此导致该例患者的α地中海贫血临床表现较为明显。
重型复合型患者的贫血症状差异较大, 从重度贫血到轻度贫血都有, 其中2例重度贫血的基因型分别是41~42/654复合-α3.7/αα和-29/654复合-α3.7/αα, 这2例患者的血红蛋白电泳结果为HbA2增高, 而无HbF。我们上面提到, α地中海贫血基因中和β地中海贫血临床表现的程度依赖于失活的α珠蛋白基因个数, 这2例患者均复合-α3.7/αα, 失活1个α基因, 不足以平衡α和β肽链之间的不平衡状态, 因此为重型β地中海贫血的临床表现。其余22例重型复合型患者, 包括1例同时合并HbH病的患者, 均表现为中度至轻度贫血, 临床上不完全依赖输血, 即中间型地中海贫血。这是因为重型β地中海贫血同时复合α地中海贫血时, β肽链合成减少的同时, α肽链亦合成减少, 二者不平衡的状态得以减轻, 因此临床症状也可因而减轻, 因此重型复合型患者是中间型地中海贫血的主要基因型组成之一。此外, 这些患者中的1例同时又复合HbH病的患者表现为中间型地中海贫血的原因也可以用上述失活的α珠蛋白基因个数理论解释, 我们之前也有相关的病例报道。而其他病例除α珠蛋白基因失活理论外, 都有一个共同特点, 即HbF的明显增高。γ珠蛋白表达增高, 与过剩的α珠蛋白形成胎儿血红蛋白HbF, 可以修正由于β珠蛋白减少造成的珠蛋白肽链比例失衡, 这一特点使HbF的表达水平成为β地中海贫血表型严重程度的重要修饰成分。β地中海贫血患者间的HbF水平相差很大, 某些重型β地中海贫血个体无HbA, 但若合并存在高表达的HbF, 可使其体内的血红蛋白维持在一定水平而无需定期输血, 呈现中间型β地中海贫血表型。
一般实验室筛查地中海贫血, 是根据红细胞脆性降低或MCV下降, 再结合HbA2和HbF水平来判定是α地中海贫血或β地中海贫血。由于地中海贫血在中国南方的高发状态, 导致αβ复合型地中海贫血患者也占有相当的比例。从我们的资料来看, 69例αβ复合型地中海贫血检测出的基因突变类型, 以中国人群中最常见类型为主, 目前市场上的地中海贫血基因诊断试剂框几乎全部覆盖了这些突变类型。也就是说, 我们在日常工作中只要稍加留心, 即可避免该类型地中海贫血的漏诊、误诊。轻型复合型患者多表现为HbA2和(或) HbF水平升高和红细胞脆性改变等β地中海贫血杂合子的特征, 而α地中海贫血的特征被掩盖, 临床上在遇到此类标本时, 尤其在孕前或产前筛查中已有一方诊断为α地中海贫血的情况下, 应对β地中海贫血杂合子同时进行α地中海贫血的基因检测, 在降低漏诊和误诊率的同时, 也对指导地中海贫血产前诊断和避免重症及中间型地中海贫血患儿的出生意义重大。
近年来, CRISPR/Cas9的问世, 在基因编辑领域中引起了革命性的变革。CRISPR/Cas9系统的基因治疗成为治愈此病的新宠。Liu等筛选出最优gRNA以及小分子化合物(L755507) , 同时联合ss ODN, 转染β-地中海贫血患者的i PSCs, 修复β41-42(TCTT) 的删除突变。应用流式细胞术(fluorescence-activated cell sorting, FACS) 检测转染细胞, 基因修正效率显著提高到79.5%, 其中等位基因修复率高达54%。脱靶检测及全外显子基因测序结果表明, 利用CRISPR/Cas9修复患者i PSC是一个安全且无缝修复方法。这对于CRISPR/Cas9日后应用于临床来说至关重要。为验证校正后i PSC的多能性, 研究者利用免疫荧光分析, 发现校正后的细胞保留了统一的多功能标记。更将校正后的i PSC细胞移入严重免疫缺陷的小鼠体内, 观察8周后, 组织学检查验证了小鼠体内的畸胎瘤, 包含所有细胞类型。更为重要的是, 从修复后的i PSC分化的祖细胞及红细胞检测证实修复后的细胞表达了完整的β-血红蛋白的mRNA。这项研究结果是在基因修复上至关重要一步, 为将可将CRISPR/Cas9系统得以应用于临床上提供原则性的理论依据。
无独有偶, 与Liu等不同, Mettananda等基于α-珠蛋白链与β-珠蛋白链不平衡所导致的无效红细胞生成和溶血的β-地中海贫血的病理生理;并了解了当α-地中海贫血合并β-地中海贫血时, 过多的游离的α-珠蛋白就会大大减少, 从而很大程度减轻患者地中海贫血的临床表现;利用CRISPR/Cas9系统在β-地中海贫血患者的人造血干细胞(CD34+) 上α-珠蛋白MCS-R2增强子进行删除突变, 形成α-地中海贫血。编辑后的CD34+细胞分化为成熟红细胞, 研究者如期观察到红细胞中α-珠蛋白表达减少, 从而纠正了α-珠蛋白链与β-珠蛋白链不平衡的病理生理, 改善了患者的临床症状。利用含有一定比例编辑后的CD34+细胞进行移植已成为治疗β-地中海贫血的有效方法。
----
中国南方α地中海贫血基因突变型
中国人非缺失型突变以Hb CS突变为主
到目前为止, 全世界已报道35种α地中海贫血缺失型突变, 其中-α4.2和-α3.7缺失型是遍布全世界的突变型, 其余突变则随地理和人群分布的不同而有差异, 这主要是由于染色体发生不等交换所致。已报道的非缺失型α地中海贫血在40种以上。我国南方各省为α地中海贫血高发区, 其分子基础主要是大片段基因缺失。--SEA, -α3.7和-α4.2为最常见的3种基因缺失类型。国内已发现的非缺失型α地中海贫血突变主要是Hb CS和Hb QS两种。除此之外, 1981年, Liang CC等在中国人中发现Hb Duan突变。1988年, Liang S等在中国人中发现Hb Westmead和α-地中海贫血-2(-4.2 kb) 一起引起的HbH病。赵永忠等也报道了在
HbH患者中发现Hb Westmead和α2CD31AGG→AAG(Arg→Lys) 突变。1990年, Zhao等在澳门的中国人中发现HbHekinan。最近在香港发现HbH-codon 30, Hb H-codon 31和Hb H-codon 59几种突变类型, 说明中国人非缺失型α地中海贫血的分子基础较复杂, 有待进一步研究阐明。
引起我国α地中海贫血的基因型主要类型依次为为东南亚缺失型(--SEA) , -α3.7, -α4.2和Hb CS。发现2例无血缘关系的Hb Westmead(α2CD122His→Gln) 导致的静止型α地中海贫血患者, 此前这种点突变国内仅在HbH病中发现。从本实验的结果分析, 此种突变在中国人中并非罕见。在对标准型和静止型α地中海贫血患者356人进行基因型检测分析中, 有51人未能确定其基因型, 这需要做特别说明。出现这种情况有两种可能:一是本教研室根据红细胞渗透脆性变化建立的“一管筛查法”筛查出地中海贫血携带者的方法不可能百分之百准确, 存在一定数量的假阳性结果;二是这些标本中应该还存在一些我们目前还未能筛查出的突变类型。所以在最后计算突变基因频率时, 我们舍去了对这51人的统计。
目前已知HbH病的基因型主要为右缺失型(--/α-α3.7) , 左缺失型(--/α-α4.2) 和非缺失型(--/αTα, ααT/αTαT) 。不同地区报道的HbH病各种基因型频率各异。我们的结果显示, 引起我国两广地区HbH病的基因型主要有右缺失型(-- SEA /α-α3.7) , 左缺失型(-- SEA/α-α4.2) 和非缺失型(-- SEA /ααCS和-- SEA /ααQS) 。其中右缺失型占37.2%, 左缺失型占25.6%;非缺失型为26.9%, 其中Hb CS占绝大多数(19/21) , 这和我室以前的报道基本相符。值得注意的是, 广东籍HbH病患者基因型多数为缺失型, 且多数为右缺失型(63例标本中, 有20例--SEA /α-α4.2, 29例--SEA /α-α3.7, 9例--SEA/ααCS, 1例--SEA/ααQS, 未知突变4例) , 而广西籍患者绝大多数为非缺失型Hb CS所致(15例标本中, 有10例--SEA/ααCS, 1例--SEA/ααQS, 未知突变4例) 。虽然广西籍HbH标本的数量及采集地有局限性, 且有报道表明广西地区HbH病缺失型以左缺失型为主。这表明两个地区的HbH病基因型分布有一定差异。在2例广西籍HbH患者的α2基因第65密码子(第二外显子) 发现一尚未见报道的同义突变。它与HbH病表型究竟有无关系, 抑或是DNA单核苷酸多态位点(SNPs) , 有待进一步研究证实。1994年, 阎志杰等在2例中国人HbH病标本中亦发现了在α2基因的第124密码子发生同义突变, 当时未能弄清它与HbH病表型究竟有无关系。同义突变是否对α珠蛋白表达过程的某些环节产生影响或是不同的密码子在编码同一个氨基酸时有效率差异, 这一现象再次在HbH病中发现应该引起我们的注意, 在功能学及基因和蛋白的关系方面值得进一步深入研究。以上发现和数据, 对于丰富我国地中海贫血的分子遗传学资料, 调整制定我国南方α地中海贫血基因诊断和产前基因诊断策略及进一步建立完善诊断方法具有现实意义。
本研究根据突变扩增系统基本原理建立了检测Hb CS和Hb QS突变的4P-ASPCR新方法。本方法能够采用2对引物分别反向扩增正常及突变等位基因, 在同一个PCR反应管中一次鉴定出Hb CS或Hb QS纯合子、杂合子及正常人。这大大增加了PCR诊断的可靠性并克服了以往ARMS法需要设计5条引物, 在2个反应管中进行反应才能完全鉴定杂合子和纯合子突变的缺点。此方法优于现已报道的方法。此设计思路亦可用于检测其它类似点突变。
众所周知, α珠蛋白基因簇中由于存在许多高度同源序列, 且GC含量高, 富含非甲基化的CpG岛及Alu重复序列, 使PCR用于α地中海贫血的基因诊断困难重重, 特别突出的是检测-α4.2和-α3.7时的非特异性扩增和重复性差的问题。本研究建立和完善的以PCR为基础的基因诊断方法基本克服了以往基因诊断方法检测α珠蛋白基因突变时普遍面临的准确性差、特异性不高或操作复杂等问题。
----
造血干细胞治疗
自1982年美国Thomas等首例报道用异基因骨髓移植成功治愈1例14月龄的重型β地中海贫血患者以来, 国外学者对造血干细胞移植进行了进一步的探索和研究。根据造血干细胞来源, 可分为骨髓移植, 外周血造血干细胞移植, 脐血移植, 子宫内骨髓移植。骨髓移植的实质是造血干细胞移植, 干细胞能够自我复制并分化为免疫活性细胞和成熟血细胞。自20世纪80年代初国外开始骨髓移植治疗重型β地中海贫血的工作以后, 不断有成功的报道。但有一个因素限制了骨髓和外周血干细胞移植的开展, 那就是血缘相关的人类白细胞抗原全相合供体来源有限。脐血干细胞拥有独特的生物学特性、资源优势以及广泛的临床适应证, 可以弥补骨髓及外周血造血干细胞移植的不足。现已证实脐血含丰富原始造血细胞, 且增殖潜能强。脐血移植的优点有:同胞脐血移植能够彻底治愈重型地中海贫血, 其移植相关死亡率低;能克服人类白细胞抗原不全相合的障碍, 与骨髓移植相比, 移植物抗宿主病发生率及程度较低;患者有机会更早接受移植治疗, 减少治疗费用;减少输血, 减轻铁过载造成的脏器损害。脐血干细胞移植让更多没有合适同胞供者的地贫患者获得移植治疗机会, 已有多例脐血移植成功用于治疗地中海贫血的报道, 但提高其临床治疗效果仍需要深入研究。
----
产前诊断
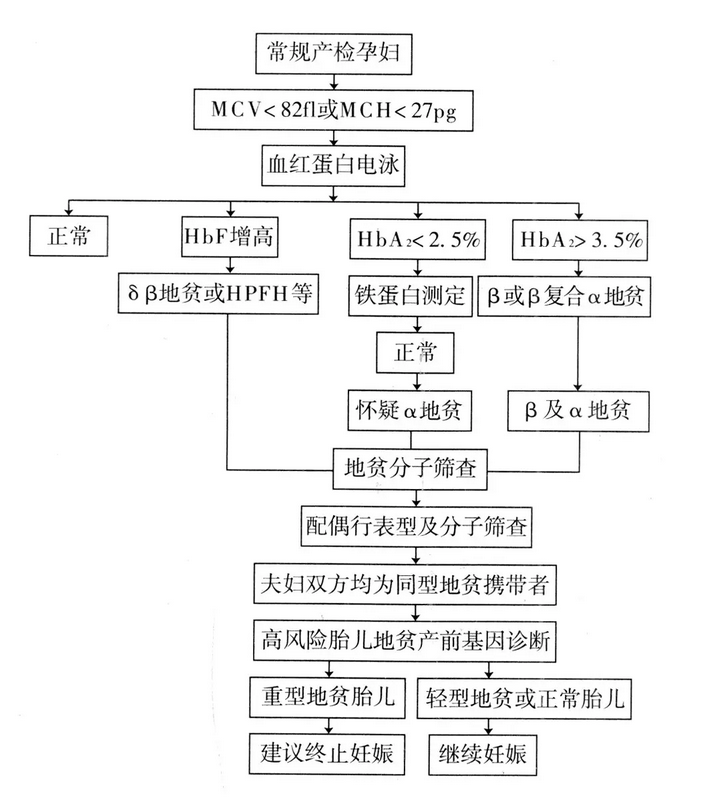
地中海贫血产前筛查及产前诊断流程
对孕期地贫筛查结果为高风险的夫妇, 医生应及时给予优生遗传学指导, 做好产前诊断, 并在严格遵循知情选择原则的前提下, 给予生育指导。
地贫产前诊断的对象
曾经分娩过重型或中间型α或β地贫患儿的夫妻;夫妻双方均为α地贫携带者, 或一方为α或β地贫携带者而配偶为β地贫复合α地贫携带者;夫妻双方均为β地贫携带者。
地贫产前诊断的时机
产前诊断宜在妊娠24周前进行。综合多篇文献的经验和建议, Eisenberg等认为, 早孕绒毛组织检查宜在妊娠10~13周进行, 羊水穿刺宜在妊娠15~18周进行。临床实践中, 根据不同患者的个体差异, 胎儿样本取材时间也可以适当调整。脐静脉穿刺检查在地贫产前诊断中较为少用。
在严格的知情同意前提下, 同时采集携带者夫妇外周血和胎儿组织(绒毛、羊水或脐血) , 分别提取DNA后, 进行基于完整家系分析的基因诊断和产前诊断。在条件允许的情况下, 产后尽可能采集到胎儿的血样或组织, 以便进一步验证产前诊断结果。
地贫的产前诊断
α地贫
α地贫的产前诊断主要是针对有Bart's水肿胎风险的夫妇, 目前针对此类夫妇, 除采集胎儿组织进行基因分析和产前诊断外, 用超声进行重型α地贫早期产前诊断也已被广泛应用。因为超声方法简单无创、阳性率高、风险性小、减少了孕妇的痛苦而越来越被人们所接受。Bart's水肿胎早期出现的最明显的超声改变为胎儿全身皮肤增厚、腹水、胎盘水肿, 随着妊娠周数的增加, 可逐渐出现肝脾肿大, 胎儿腹围异常增大, 并出现心脏增大、心胸横径比增大、心室壁增厚、胸腔积液、心包积液等胎儿心功能不全的超声声像图改变。但该方法对观察者的技术及设备要求较高, 且特异性较差, 任何原因引起的胎儿水肿均可显示类同Hb Bart's水肿胎的声像图改变, 如胎儿宫内感染、双胎输血综合征、心脏肿瘤等。如果能结合相关检查, 有助早期发现和明确诊断。
此类夫妇的遗传咨询中应告知产前筛查和产前诊断的必要性, 取得孕妇及家人的理解。对于产前诊断结果为Bart's水肿胎的家系, 则需要向患者说明可能实施终止妊娠的操作程序, 应使其在1周内终止妊娠, 以避免不必要的身体和精神伤害。
HbH病的产前诊断和选择性流产尚未被公认, 故其医学实践需慎重考虑。鉴于一些严重类型的HbH病患者(如- -SEA/αCSα) 需依赖输血和去铁治疗, 生存质量明显下降, 当夫妇面临其胎儿有这种疾病风险, 要求产前诊断且同意接受对HbH病胎儿的终止妊娠操作时, 其选择应得到尊重。
β地贫
产前诊断结果为重型β地贫胎儿的家系, 其遗传咨询原则同重型α地贫。
对于中间型β地贫的遗传咨询, 也基本与HbH病的咨询原则相同。但因中间型β地贫的临床异质性更为复杂, 在该病的遗传咨询过程中, 更需要慎重和严格的知情同意。
----
DNA甲基化
近年来, 国内外表观遗传学的相关研究已经逐步开展, 并取得了相当大的进展。这其中, DNA甲基化是表观遗传学中研究最深入的一种。DNA甲基化是哺乳动物基因组的显著特征, 它是在DNA甲基转移酶(DNA methyltransferase, DNMT) 作用下, 以S腺苷甲硫氨酸为甲基供体, 将甲基转移至胞嘧啶的5′位置上, 形成5-甲基胞嘧啶(5-mC) 。DNA甲基化是细胞开闭基因表现的一种方式, 基因表达水平与DNA甲基化水平呈负相关, 尤其是启动子区域的低甲基化和转录激活直接相关。研究表明, DNA甲基化成为除DNA突变和缺失以外导致基因失活的另一重要机制。DNA甲基化引起基因突变的机制主要是由于DNMT催化反应形成。DNMT可以加快胞嘧啶(C) 和5-mC脱氨, 封闭尿嘧啶(U) 修复, 并且使U→T改变, 故DNMT促使CpG序列的C→T突变。总之, DNA甲基化改变可直接影响基因转录, 通过5-胞嘧啶甲基化改变, 引起基因表达异常, 但DNA顺序和基因产物不变;也可通过5-甲基胞嘧啶去NH3诱发C→T(胸腺嘧啶) 突变而影响基因表达。
目前许多血液病与表观遗传学的相关研究在国内外已经展开。白血病、淋巴瘤、骨髓增生异常综合征及浆细胞疾病等的研究已经逐渐开展, 而目前有关地中海贫血的表观遗传学发病机制的相关研究正处于起步阶段。有研究报道, 表观遗传学机制可能是包括地中海贫血在内的人类遗传性疾病的发病机制之一。目前, 地中海贫血的表观遗传学研究国外只有极少数的相关报道, 而国内尚未发现有相关文献报道。
几十年来多方面的研究证明, 从细胞系到鼠、鸡、兔以至灵长类和人, DNA甲基化修饰与类珠蛋白基因表达调控有关。从有关人类β珠蛋白基因簇ε、γ、β各基因在个体发育不同阶段的DNA甲基化模式研究发现, 当ε、γ、β基因分别在卵黄囊、胎儿肝脏、成人骨髓造血阶段表达时, 各表达基因的启动子区DNA呈低甲基化状态, 其他沉默基因启动子区DNA呈高甲基化状态。国外另有动物实验研究报道, 和人类有类似的珠蛋白基因开关的狒狒的基因表达和珠蛋白启动子甲基化有关。
Tufarell等研究发现一种α地中海贫血患者, 虽然α珠蛋白基因缺失, 但与之相邻的LUC7L基因缺失了一大段DNA。当细胞以这种突变型的DNA为模板转录RNA时, 产生了一种带有部分LUC7L序列的RNA, 但这种RNA的一部分也与α珠蛋白基因互补, 叫反义RNA。α珠蛋白的这种反义RNA导致正常HBA2基因发生甲基化而沉默, 使得正常基因失去功能, α珠蛋白链发生缺失。
----
组蛋白修饰
组蛋白修饰较DNA甲基化修饰复杂很多, 因为不同组蛋白(组蛋白H3和H4) 的不同氨基酸序列(H3末端有7个Lys和2个Ser;H4末端有5个Lys和一个Ser) 可发生不同类型的修饰, 包括乙酰化、甲基化、磷酸化、泛素化、糖基化等, 它们都是组蛋白密码的基本元素。其中最多的是乙酰化修饰。
组蛋白乙酰基转移酶将乙酰辅酶A的乙酰基转移到核心组蛋白氨基末端, 氨基的正电荷被消除, 从而有利于DNA构象的展开, 使核小体的结构变松弛, 进而促进转录因子和辅助转录因子与DNA分子的接触, 而可激活特定基因的转录过程。组蛋白去乙酰化酶则移去组蛋白Lys残基上的乙酰基, 恢复组蛋白的正电性, 带正电荷的Lys残基与DNA电性相反, 增加了DNA与组蛋白间的吸引力, 使启动子不易接近转录调控元件, 从而抑制转录。
对人类β珠蛋白基因簇组蛋白修饰模式的研究显示, β珠蛋白基因在不同发育阶段的沉默或激活随组蛋白乙酰化水平的变化而改变, 即组蛋白乙酰化水平增高, 基因激活, 降低则基因沉默。对表达胎儿γ珠蛋白基因的K562细胞表观遗传修饰分析显示, 从基因座位调控区到胚胎、胎儿基因都存在广泛乙酰化, 在沉默的β基因缺乏H3乙酰化。目前许多有关组蛋白乙酰化酶抑制剂, 如曲古抑菌素、丁酸盐等, 诱导血红蛋白F的实验及临床研究观察到血红蛋白F增加伴有γ珠蛋白基因启动子区组蛋白呈高乙酰化状态。另外, 有研究表明GATA结合蛋白1、红系核因子2、红系Kruppel样因子等红系特异转录因子与β珠蛋白基因的表观遗传修饰有关。
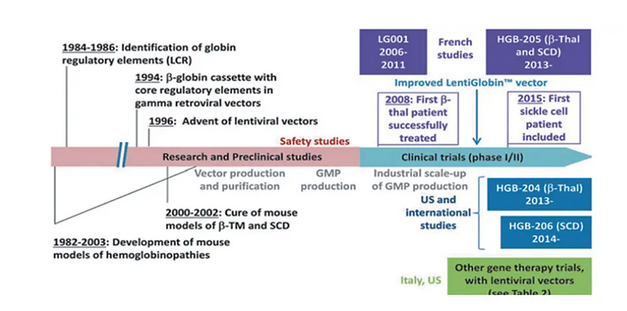
地贫基因治疗研究进展及重要突破
因治疗的主要步骤示意
用于地贫基因治疗的病毒和载体基因组结构示意
----
地贫的基因治疗
2.1 基因治疗的可行性
地贫的分子机制主要涉及珠蛋白基因的改变而导致血红蛋白α链与β链不能形成正常的Hb A四聚体, 从而产生异常血红蛋白, 通常异常结构的血红蛋白容易受到氧化损伤, 所以患者临床主要表现为溶血性贫血和携氧能力降低。基因治疗技术对单基因突变引起的疾病有较好的治疗效果, 因为地贫是单基因隐性遗传病, 所以基因治疗技术是目前地贫治疗研究的热点。基因治疗的常规技术手段是将正常的目的基因片段通过表达载体导入到受体细胞中, 并在受体细胞中成功表达, 从而恢复有缺陷基因的功能。
在人的不同发育阶段珠蛋白基因的表达和调节能够组建正常的珠蛋白, 而这个表达和调节过程十分复杂。在β珠蛋白基因上游有一段40~60 kb片段被称之为长末端重复序列(long terminal repeat, LTR) , 它包含有特异性增强子序列, 能够高水平表达β珠蛋白基因, LTR区域包含有5个对DNA酶敏感的位点:HS1至HS5, 在5个HS区域中, HS2对LCR的孤立表达是至关重要的, 而HS3能够对其增强子活性有重要的阻碍作用, 通过这些位点调控能够影响β珠蛋白的表达量;而类似于β-珠蛋白基因的LCR区域, 在α珠蛋白基因上游的40 kb区域发现一段具有增强子作用的位点HS40, 该位点也同样对DNA酶敏感, 具有可调控性。所以对珠蛋白基因进行基因编辑和调控以恢复珠蛋白的正常生理功能, 在理论上是可以治愈地贫的。1982-2015年地贫基因治疗的研究不断突破(图1) , 各国的研究报道证明对地贫的基因治疗是可行的。而成功进行血红蛋白疾病基因治疗需要满足以下要求:(1) 高效率的目的基因转移和高比率的造血干细胞(hematopoietic stem cells, HSC) 植入;(2) 移植位置孤立并且转导HSC能够一致性表达;(3) 能够高水平表达β-/γ-珠蛋白基因;(4) 少量或没有插入突变和肿瘤发生风险;(5) 红细胞谱系特异性以及在发育阶段特异性表达的转移β-珠蛋白基因检测手段。
基因治疗的主要步骤(图2) :(1) 分离并收集自体粒细胞集落刺激因子(granulocyte colony-stimulating factor, G-CSF) 动员后的纯种外周血HSC或骨髓HSC;(2) 用基因治疗载体转导HSC 24~48 h;(3) 给予患者化疗, 消除体内所剩存的HSC, 为移植转导后的HSC做准备;(4) 基因转导的HSC通过静脉输注进入体内;(5) 患者体内珠蛋白基因表达水平监测以及生存质量维护。世界上第一例应用基因治疗技术治疗地贫患者并取得成功是在2007年, 患者的转导效率能够达到30%, 虽然在进行基因治疗的第一年只有有限的珠蛋白表达, 但两年后由于转基因表达逐渐完善, 患者的血红蛋白检测稳定在8.5~9 g/d L范围内。
在所有的基因治疗研究中我们不难发现, 有效的病毒载体和骨髓干细胞移植是基因治疗的关键所在。
2.2 基因治疗中的病毒载体
临床前研究和临床试验中的大多数病毒载体来源于逆转录病毒科, 其他病毒载体包括腺病毒、单纯疱疹病毒、牛痘病毒、痘病毒等都缺乏整合装置, 而逆转录病毒却可以永久整合到细胞基因组中。目前用于临床试验或进行临床前检测的逆转录病毒科病毒主要有逆转录病毒(retrovirus, RV) 和慢病毒(lentivirus, LV) 。通过生物工程等相关技术将这些病毒载体颗粒除去致病性和自身复制有关的基因元件, 并用目的基因转基因取代, 利用这些病毒载体可以将正常的珠蛋白基因整合到人类基因组中, 从而纠正潜在的遗传缺陷(图3) 。
用于基因治疗的初始载体来源于小鼠莫洛尼白血病病毒(moloney leukemia virus, MLV) , 它是一种简单的逆转录病毒, 属于γ-逆转录病毒家族, 被称为RV载体, 而用于基因治疗的理想靶细胞通常是HSC, 因为HSC从骨髓中能比较容易地分离出来, 并且能动员造血, 同时骨髓移植技术和细胞培养技术已经发展成熟, 所以利用RV具有将RNA逆转录成互补的c DNA特性, 可以将c DNA整合到宿主细胞基因组中以递送目的基因进入细胞达到基因治疗的目的。但RV载体也有一定的局限性, 首先它不能感染处于静息状态下的HSC(不进行分裂的HSC) , 这就可能使目的基因不能导入靶细胞, 目的基因不能成功在HSC中表达, 同时RV载体能编码β-珠蛋白相关基因, 在LTR区域含有珠蛋白基因的启动子和增强子, 有一定概率能够整合到原癌基因附近造成肿瘤的发生。对于血红蛋白类疾病, 还有一个额外的限制因素就是RV载体无法携带大量的珠蛋白基因, 且对载体监测需要在其高水平表达后才能监测。这些限制促使人类免疫缺陷病毒(human immunodeficiency virus, HIV) 载体的发展。艾滋病属于LV家族的逆转录病毒, 因此这些载体被称为LV载体。LV对于血红蛋白病的基因治疗有独特的几个优点:(1) LV具有进入并融入非分裂细胞完整核的能力, 因此它们能有效地转导HSC;(2) LV可以被设计成具有“自我失活”能力的“装置”, 当启动“自我灭活”时, 所有的具有转录作用的病毒都将失去效应;(3) LV具有携带大量的基因能力, 所以非常适合地贫等血红蛋白类疾病的基因治疗。应用LV载体进行小鼠地贫基因纠正模型最早由May等报道, 通过基因治疗后发现每3~4 g/d L拷贝的LV载体能使地贫小鼠的平均血红蛋白升高, 红细胞指数得到校正。
利用病毒将正常的β珠蛋白基因导入人体内, 并在人体内成功表达β珠蛋白, 最终使β地贫患者获得终生的治疗, 这不仅证明基因治疗方法能够有效地治疗地贫, 同时也为地贫治疗领域开启了新的一页, 奠定了基因治疗在临床上的重要运用的基础。Gambari采用病毒和非病毒DNA方法治疗β-地中海贫血策略, β-珠蛋白基因表达修饰可以通过突变的β-珠蛋白基因碱基置换和RNA修复来实现, 同时基于增加胎儿血红蛋白(Hb F) 合成对β-地中海贫血有益的概念, 已经优化了通过DNA方法来增加Hb F的生成量, 包括用携带γ珠蛋白基因的病毒载体处理靶细胞, 使γ珠蛋白的表达量成倍增加等。
2.3 同种异体造血干细胞移植治疗
目前异体造血干细胞移植(allogeneic hematopoietic stem cell transplantation, allo HSCT) 是唯一已经建立完全, 并且是针对β地贫和镰状细胞贫血(sickle cell disease, SCD) 患者的确切治疗方案。在这两种贫血中, 儿童群体匹配同胞供体(matched sibling donor, MSD) allo HSCT总体生存率表现优异, 生存率大于80%, 移植失败率仅10%。然而, 大多数患者并没有人类白细胞抗原(human leucocyte antigen, HLA) 匹配合适的同胞。目前可匹配的无关供体(matched unrelated donor, MUD) 的allo HSCT越来越多地被用来治疗地贫和SCD, 但研究发现只有不到14%的人有一个匹配的兄弟姐妹, 而在美国约30%的非洲裔美国人才能有一个MUD。有关数据显示, 在移植低风险群体中, 移植后的地贫患者生存率为80%。然而高风险群体(如肝脾肿大者) 的移植结果却不容乐观, 移植后的生存率相对比较低, 生存率约为54%。经调查研究这些死亡率大多归因于二级至四级移植物抗宿主疾病(graft-versus-host disease, GVHD) 或移植物衰竭。
同时异体基因间患者的HSCT治疗还面临着许多问题, 因为allo HSCT会产生许多相关的副作用导致异体基因不能被受体接受, 此外接收治疗之前的大多数患者往往都接受过输血治疗, 重复的输血治疗导致这些受试者的肝脏和免疫系统活动性增加, 会增加移植后排斥反应, 降低患者的生存率, 所以受试者在进行allo HSCT之前需要给予白消安和环磷酰胺等药物进行调理, 以最大程度的降低移植风险。在过去十年的研究发展中, allo HSCT移植结果已经得到明显的改善, 因为已经形成一套合理的方案来辅助allo HSCT。首先通过风险分层筛选出高低风险群体, 评估受试对象的风险值, 再根据不同对象给予合适的白消安靶向剂量, 并在调理期间根据患者实际情况不断改良调理方案和改善支持性护理。
在异体干细胞移植前也可以通过靶向核酸酶的基因组编辑技术对其进行基因编辑, 构建正常且完整的珠蛋白基因组。近期研究热门的CRISPR Cas9基因编辑系统可以实现对HSC的基因编辑, 相对于锌指核酸酶(zinc fingers nucleases, ZFN) 等技术, 它具有非常强大的基因编辑功能, 也更安全、更有效。进行基因编辑后的HSCT必须要获得大量基因编辑成功的HSCs, 只有这样才能保证在移植后能够产生所需的血红蛋白量, 但到目前为止用基因编辑方法在地贫治疗中对HSCs进行靶向基因修正, 其实际效率往往是低下的。Hoban等和Chang等使用一种关于ZFN技术的改进方法, 能使基因编辑效率显著提高。这些基因编辑技术无疑给HSCT的发展创造了更多的可选择性, 也使转化效率得到进一步提高。
----
地中海贫血诊治情况及国内现
血红蛋白病流行病学调查与我国现状
2008年WHO全球血红蛋白病流行病学报告:在229个国家中约占71%人群存在血红蛋白病这一重大健康问题。在这些国家出生的新生儿占全球新生儿的89%, 其中每年超过330 000例的新生儿与血红蛋白病有关(其中83%为镰状细胞症, 17%为地中海贫血) 。5岁以下儿童死亡者中血红蛋白病约占3.4%。每年约有56 000例重型β-地中海贫血, 其中至少有30 000例需规范输血才能存活, 5500例重型α-地中海贫血死于围生期。
20世纪80年代曾溢滔院士主持开展了我国20省(市) 自治区近60万人口的地中海贫血流行病学调查工作。根据十二省地中海贫血的流调结果, 该病主要分布在我国长江沿岸及以南的地区, 包括11个省、直辖市和自治区, 以及沿丝绸之路分布的陕西、甘肃、新疆等地。按当时的流调结果, 该病的检出率为3.62%, 其中α和β地中海贫血的检出率分别为2.95%及0.67%。
近30年来未再开展全国范围内本病的流行病学调查工作, 只有广东、广西、云南等省开展区域性的流调工作。我国南方地区各地报道的地中海贫血基因缺陷率为2.5%~20%, 而广东及广西两省地中海贫血基因缺陷发生率分别高达10%及20%, 两省地中海贫血的病例数占全国总数的2/5以上。若按平均3%的β地中海贫血基因携带率估算, 广东省每年出生重型B地中海贫血新生儿约500例, 10年累计约5000例。因此, 地中海贫血已经成为广东等高发地区社会性公共卫生问题。
我国地中海贫血发病率较高的地区还有海南、贵州、云南、四川、重庆、福建、湖南、湖北、江西等省市。但在近年有关地中海贫血发病率的文献报道中, 提示还有相当多的地区未开展地中海贫血的流行病学研究。由于缺乏全国流行病学资料, 影响国家与地方政府对地中海贫血防治的重视, 未能从国家与地区层面制定相应的防控措施。因此, 有必要开展全国或高发地区血红蛋白病的流调工作, 为国家或地区制定血红蛋白病的防治政策, 建立防控网络组织提供科学依据, 最终达到降低重型地中海贫血患儿的出生率, 从根本上减轻家庭与社会的压力和负担。
地中海贫血防治与我国现状
1987年国际地中海贫血联盟(Thalassemia Internatinal Federation, TIF) 成立, 该组织旨在为全球受血红蛋白病影响的国家提供有效的国家地中海贫血预防及管理方案。目前TIF已有60个国家的98个国家地中海贫血组织参加。
1996年TIF正式与WHO合作, 来自全球60多个国家的科学家与医学专家, 还包括国际卫生机构、医药公司及病患组织, 形成了广泛联盟的合作网络。TIF执行总监Androulla Eleftherious指出:健康权威专家们需要认识到——血红蛋白病是一个值得重视的社会公共健康问题, 应从国家层面上制定政策, 以促进和改善血红蛋白病的诊疗与预防现状。支持政策应包括:(1) 制定实验室血红蛋白病诊断标准指南;(2) 制定全国性地中海贫血诊疗指南;(3) 地中海贫血流行病学资料与监测;(4) 建立针对卫生专业人员、患儿及其父母和公众的地中海贫血教育计划。
随着计算机与互联网技术的普及与发展, 一个构建于国家层面的全球性血红蛋白病流行病学资料数据库已在一些国家成立。并利用流行病学的方法和数据, 评估遗传性血红蛋白病的治疗和预防, 论证其服务需求的公平性和成本效益的研究。
我国已在广东及广西等省建立了较为完善的地中海贫血筛查预防网络, 将地中海贫血的筛查工作纳入在各市、区级医院和妇幼保健院对孕妇常规检查项目, 为防止重型地中海贫血出生做出了成绩。
但地中海贫血在我国南方分布广泛, 有关地中海贫血的防治知识尚不普及, 各省尚未将地中海贫血筛查纳入婚前和孕妇常规检查项目。基层医务人员对该病认知水平不高。尤其是近年取消了强制性婚检制度, 导致各类出生缺陷率明显上升, 每年仍有不少重型地中海贫血患儿出生。仅以深圳市儿童医院为例, 2006年新诊断重型β地中海贫血12例, 2007年9例。这些患儿父母对本病不了解, 多系未进行过相关地中海贫血的婚前及产前检查的外来务工人员。
2010年, 中华医学会儿科学分会血液学组制定了重型β地中海贫血的诊断和治疗指南。同时, 建议各地根据本地区实际情况, 开展地区性的流调工作, 了解本地区地中海贫血的基因携带率、重型β-地中海贫血发病率和特征。为国家、地方政府制定相关政策提供科学依据。
与地中海贫血防治先进的国家相比, 我国地中海贫血防治工作任重道远。目前尚未建立覆盖全国的地中海贫血管理组织和防控监测网络;数十年来未开展血红蛋白病的流行病学调查, 更谈不上建立全国性地中海贫血流行病学资料数据库。因此, 呼吁建立一个由政府主导, 由中华医学会儿科分会血液专业组牵头, 计划生育、妇幼保健机构及地中海贫血患儿家长联盟共同参与的地中海贫血防控组织十分必要, 以促成地中海贫血高发地区尽快建立信息化完善的地中海贫血监测网络。
地域分布
地中海贫血诊治进展与我国现状
诊断进展
目前发达国家对地中海贫血的诊断与筛查主要运用基因诊断。Old等总结了英国25年来血红蛋白病产前诊断的准确性研究与经验总结。回顾自1974年以来英国居民进行地中海贫血和镰状细胞病产前诊断。3254例行妊娠产前筛查:其中517例进行胎儿血液分析, 681例应用DNA印记法(Southern blotting) , 2056例做聚合酶链反应(PCR) 检测, 半数以上使用基因扩增技术(amplification refractory mutation system, ARMS) 。确诊纯合子808例, 占24.8%。25例诊断错误, 其中, 10例非实验室错误, 占0.31%, 15例是与实验诊断相关的技术问题。数据显示产前诊断的准确性随着每项诊断技术的发展而改善, 可以肯定ARMS-PCR技术对β-地中海贫血和镰状细胞病产前诊断的准确性和可靠性。目前PCR方法在英国产前诊断的总体错误率是0.41%。
由于我国各级医院开展地中海贫血实验室诊断条件尚不完善, 诊断等级可分为:(1) 初步诊断:小细胞低色素性贫血, 血常规:Hb降低, MCV、MCH、MCHC降低;(2) 重型β地贫Hb F明显升高, 可达30%~90%;中间型β地贫Hb F>3.5%;轻型β地贫Hb A2>3.5%或正常, Hb F正常或轻度增加(不超过5%) ;重型α地贫Hb Bart成分>70%, 少量Hb Portland, 可出现微量Hb H;中间型α地贫出现Hb H区带, Hb H成分占5%~30%(个别患者Hb H成分可小于5%或高达40%) , 也可出现少量Hb Bart(出生时Hb Bart可达15%以上) ;轻型α地贫血红蛋白电泳正常, 可检出ξ珠蛋白链;排除其他疾病;(3) 基因诊断:珠蛋白变异确诊。
临床分离珠蛋白肽链的常规方法主要是醋酸纤膜性电泳法, 可定量分析血红蛋白, 但分离珠蛋白肽链耗时长, 操作繁杂, 定量精度差。高效液相色谱(HPLC) 法分离珠蛋白肽链分辨力强, 准确省时, 但试剂用量大, 成本较高, 不适于临床应用。而近年问世的毛细管电泳技术综合了电泳和色谱两者的优点。廖云星等采用改良的非涂层电泳条件进行毛细管区带电泳, 分离珠蛋白肽链。实验在40 min内, 一次性将α、β、Gγ、δ、Aγ等5种珠蛋白肽链完全清楚地分离。样品用量少(相当于0.04~0.06m L血液) , 操作简便, 重复性好, 省时, 高效。适用于临床上血红蛋白病的快速诊断。
南宁市在地中海贫血普查中用全自动毛细管电泳仪测定5088例标本, 发现异常1042例, 其中β地中海贫血757例(Hb A2>4.0%) , Hb F增高23例, 异常Hb 262例。229例同时进行毛细血管电泳与地中海贫血基因检测比较, 与β地中海贫血符合率100%, 与Hb H病和Hb CS的符合率分别是97.83%和95.00%。
治疗进展
目前国内外对地中海贫血治疗分类
(1) 主要方法:规范性长期输血和去铁治疗;(2) 根治方法:HLA相合的造血干细胞移植;(3) 姑息方法:脾切除术等。
TIF地中海贫血临床处理指南内容
输血目的:对地中海贫血患者而言, 适当的输血标准和安全输血是常规治疗方案的关键。强调在重型地中海贫血患者输血治疗前须明确5个问题:(1) 何时开始对患者进行输血治疗;(2) 什么是有效而安全的输血疗法;(3) 如何确定有效输血的血红蛋白最佳标准;(4) 必要的输血对去铁治疗的影响如何;(5) 严重的输血反应有哪些常见与少见类型。
TIF推荐使用的血液制品
(1) 去白细胞血液:多采用滤过法去白细胞。要求重型β-地中海贫血应接受每袋去白细胞血液血红细胞不得低于40 g。白细胞低于每单位1×106是一个关键门槛, 该标准被认为既可消除白细胞的副作用, 又可预防输入血小板导致自身免疫反应(2) 洗涤红细胞:对于地中海贫血患者减少严重的输血反应可能有益。盐水可洗涤献血者血浆蛋白中含有针对受者的抗体。洗涤红细胞一般不能去除白细胞, 不应替代去白细胞血液制品, 相反洗涤应结合使用过滤产品。另外, 洗涤红细胞可能去除红细胞, 因此, 检查输血后血红蛋白水平, 确保目标血红蛋白水平的实现是关键。
输血计划(transfusion programmes)
治疗重型地中海贫血推荐制定规范的终身输血计划。通常每2~5周, 维持输血前血红蛋白(Hb) 在90~105 g/L以上。这样的输血计划才能保障和促进大多数患者正常生长和生理活动, 有效减少骨髓造血, 并将输血后铁负荷降到最低水平。
更高目标, 输血前Hb在110~120 g/L这一标准, 可能更适合有心脏病或医疗条件受限者, 对这些患者不需令其达到抑制骨髓造血的最低Hb水平。
重型β地中海贫血的诊断和治疗指南
系于2010年由中华医学会儿科学分会血液学组制定, 该指南参考国际地中海贫血联盟(11F) 的最新诊疗建议和有关文献, 旨在为我国儿科医生规范化诊断和治疗重型β-地中海贫血提供参考。
地中海贫血的诊断
(1) 临床表现;典型的临床特征(2) 血液学改变:(1) 外周血血红蛋白(Hb) <60 g/L, 呈小细胞低色素性贫血。出现靶形红细胞和红细胞碎片;(2) 骨髓改变;红细胞系呈增生明显活跃;(3) 红细胞脆性明显降低;(4) 首诊时血红蛋白电泳显示Hb F显著增高, 一般达30%~90%, 这是诊断重型B地中海贫血的重要依据。如Hb F不增高应排除近期输血的影响, 可在输血后3个月复查(3) 区域及家系调查:区域调查示患儿来自地中海贫血高发区域。患儿父母外周血象呈小细胞低色素性贫血, 血红蛋白电泳呈Hb A2含量升高(3.5%~6.0%)(4) 基因诊断:有条件者应进行基因诊断。可采用限制性内切酶片段长度多态性(RFLP) 连锁分析、PCR.限制酶切法、PCR.ASO点杂交、反向点杂交(RDB) 和DNA测序等方法检测β地中海贫血基因缺陷的类型和位点。
地中海贫血的治疗
规范性长期输血和去铁治疗是最主要治疗方法, 如有HLA相合的同胞供者可选择接受造血干细胞移植, 脾切除术则为姑息治疗手段(1) 输血疗法:已确诊为重型β地中海贫血患儿, 推荐:(1) Hb<90 g/L时启动输血计划;(2) 每2~5周输血1次, 每次输浓缩红细胞0.5~1单位/10 kg(我国将200 m L全血中提取的浓缩红细胞定义为l单位) , 每次输血时间大于3~4 h;(3) 输血后Hb维持在90~140 g/L。选择血液制品的原则:推荐使用去除白细胞的浓缩红细胞制品;对有严重过敏反应者应选择洗涤红细胞;避免应用亲属的血液(2) 去铁治疗(1) 铁负荷评估:检测血清铁蛋白是反映机体铁负荷状况最简单实用的方法, 建议每3~6个月动态检测1次(2) 开始去铁治疗时机和监测:输血次数≥10~20次, 或血清铁蛋白>1000μg/L。去铁治疗后每3~6个月监测血清铁蛋白, 当血清铁蛋白<1000/L可暂停使用铁螯合剂(3) 去铁药物:目前临床上应用的铁螫合剂主要包括去铁胺(desferrioxamine, DFO) 、去铁酮(deferiprone, DFP) 和地拉罗司(deferasirox, DFX, ICL670) 。
地中海贫血儿童医疗保险与我国现状
国外因实行全民医保, 或国家对弱势群体(老人、儿童、病残或失业者) 实行基本医保, 地中海贫血患者和家庭很少担心和考虑自己的医疗费用。医师在治疗患者时, 根据病情考虑诊疗方法, 很少有论文涉及地中海贫血的医疗费用问题。
英国由国家健康研究院(NHS) 制定镰状细胞和地中海贫血筛查指南, 按指南对孕妇进行产前筛查, 对新生儿进行筛查和随访检查。如在1岁内来自遗传病高发区的婴儿到英国后必须进行镰状细胞病的筛查, 费用由国家医保承担。新加坡和我国香港由于政府实施孕妇地中海贫血产前筛查, 重症地中海贫血患儿的出生率几乎为零。
美国Grosse等利用数据管理程序进行了血红蛋白病健康服务文献研究。医保数据管理程序提供了一个研究镰状细胞贫血(SCD) 或地中海贫血患者医保利用的独特方法。具有处理大样本和低成本能力, 可使临床基本数据大利用化。降低了管理数据潜在问题, 如分类不明、统计漏报、以及缺乏社会人口统计信息。引用了Delea 2008发表的论文, 研究1997至2004年间19 147例镰状细胞病(SCD) 和15 965例地中海贫血患者的输血相关资料, 有关于102例SCD和43例地中海贫血患者的陈述:因反复输血导致铁负荷增加, 去铁治疗是主要花费。
我国目前儿童的基本医疗保险尚不完善, 地中海贫血仅能报销部分医疗费用, “看病贵”对地中海贫血家庭而言仍是沉重的经济与精神负担。
2012年5月8日是第19个“世界地中海贫血日”。我国各地地中海贫血患者和医务工作者组织了活动, 引导社会关注地中海贫血患儿的医疗与生存现状, 呼吁建立完善的基本医疗保障政策, 减轻地中海贫血患者的医疗与经济负担。
----
诊断方法汇总
2.1 筛查试验
2.1.1 红细胞形态分析
地中海贫血患者血涂片检查示红细胞大小不均,常可见形态不规则的异形红细胞:如嗜碱性点彩红细胞、泪滴形红细胞、靶形红细胞等。其中靶形红细胞最多见,此种红细胞中央部位染色较深,周围区域苍白,而细胞边缘处又深染,形如射击之靶,当出现靶形红细胞时需高度警惕地中海贫血。
2.1.2 血细胞分析
红细胞基本参数是临床初步筛查地中海贫血最简单经济的常用指标 。红细胞(RBC)、血红蛋白(Hb)、红细胞压积(HCT)、红细胞平均体积(MCV)、红细胞平均血红蛋白量(MCH)、红细胞平均血红蛋白浓度(MCHC)、红细胞体积分布宽度(RDW)等参数都有助于地中海贫血的诊断。其红细胞呈典型小细胞、低色素性且大多红细胞内可见包涵体,国际地中海贫血协会推荐将MCV<78 fl、MCH<27 Pg作为筛查地中海贫血的截断值 。同时其余参数如RBC、Hb、HCT、MCHC、RDW也均有不同程度的改变,且网织红细胞比例常高于正常人。
2.1.3 红细胞渗透脆性实验
红细胞渗透脆性实验可检测在不同低渗透压盐水中红细胞的抵抗力。地中海贫血患者红细胞存在不同程度变形且体积变小,其表面积与体积的比值增大,故在低渗盐水中吸水膨胀的适应性较大(脆性降低)。但是红细胞渗透脆性实验并不能单独用于地中海贫血的诊断,必须将它和上述多种指标联合起来进行检测分析才能初步诊断地中海贫血。有学者将MCV、RDW以及红细胞渗透脆性实验联合起来用于地中海贫血的检测,可大大提高筛查的灵敏度和特异度,甚至能初步和缺铁性贫血进行鉴别 。
2.1.4 血红蛋白组分析
对血红蛋白进行分离鉴定和定量分析是诊断地中海贫血快速可靠的方法。血红蛋白组分分析能够定量检测Hb A、Hb A2、Hb F、Hb H、Hb Bart,s等,常用的分析方法有:(1)普通血红蛋白电泳法:因PH8.5缓冲盐液中各种Hb等位点均<7,在电场作用下不同的血红蛋白以不同的速度向阳极移动,常用醋酸纤维素薄膜作为支撑物来定量分析血红蛋白 (2)琼脂糖凝胶电泳:碱性环境下血红蛋白在电场中分离后染色,可对血红蛋白琼脂糖定量(3)高效液相色谱:利用离子交换树脂作为固定相,高压下血红蛋白理化不同,故在分离柱停留时间不等,从而将血红蛋白分离出来。其中,高效液相色谱分析技术对β地中海贫血、Hb变异体、Hb A及Hb F筛查的敏感度及精确度尤其高 。
目前,国际地中海贫血协会推荐将Hb A2的含量检测作为β-地中海贫血携带者诊断标准,因高效液相色谱可以同时对Hb A2进行定量分析,故广泛应用于β-地中海贫血携带者的筛查(4)毛细管电泳:血红蛋白经过毛细管分离通道,在电场中可以定向移动,不同血红蛋白移动速度不同,以此来分离各种血红蛋白并且可直接对血红蛋白进行定量分析,已逐渐在临床广泛应用。有报道称毛细管电泳综合了高效液相色谱和凝胶电泳的共同优点,具备高效、高灵敏度、可重复性等优点 。
普通血红蛋白电泳法及琼脂糖凝胶电泳因操作简单、成本低廉,故常用于基层医院地中海贫血的常规筛查;而毛细管电泳及高效液相色谱可以进一步完善前两者方法的不足,但因其成本高并且对质控要求也较高,故常用于大型实验室。
2.2 基因检测
多年的研究证实,地中海贫血是由于珠蛋白基因的缺失或点突变引起的珠蛋白链合成障碍,故针对突变基因的检测能够更加准确地诊断该病。早在1976年,已提出利用DNA-DNA核糖杂交技术检测α珠蛋白的基因,标志着基因分子诊断的诞生。随着1985年具划时代意义的聚合酶链反应(PCR)技术的发明以及近几十年来PCR相关技术的不断完善,地中海贫血的基因诊断技术得到极大的发展并成为其诊断的金标准。
2.2.1 Southern印迹杂交
该方法的原理是提取基因组DNA,用琼脂糖凝胶电泳分离经限制性内切酶消化的DNA片段,再转移到硝酸纤维膜上,最后与放射性核素标记的DNA或RNA探针杂交,用放射自显影检测酶解DNA片段的大小和位置。该方法操作所需样本量大且步骤繁琐,故其在临床中的应用受到限制。但需要提及的是Southern印迹杂交是分析大片段缺失特别是α珠蛋白基因缺陷的金标准,常作为其他基因诊断及PCR产前诊断结果的确诊试验 。
2.2.2 多重PCR及多重断裂点PCR(多重gap-PCR)
PCR原理即利用DNA聚合酶对引物进行延伸,经变形、退火、延伸过程实现特定DNA片段的体外扩增。该方法操作简便,适合基层医院对地中海贫血的临床诊断。近年来,科学家们在PCR基础上发展出多重PCR,即在同一PCR反应体系里加上两对以上引物,同时扩增出多个核酸片段。这样可以根据每个突变点特异性扩增产物来判断结果,大大提高缺失型α地中海贫血筛查(如东南亚缺失-SEA、左缺失-α4.2、右缺失-α3.7)及静止性α地中海贫血诊断。
多重断裂点PCR(多重gap-PCR)的原理是在基因缺失区域内设计两到三对引物,根据PCR特异性扩增片段有无来判断基因型,同时还可根据阳性扩增片段及正常对照片段来综合诊断被检者是地中海贫血纯合子还是杂合子或者是正常人。目前,该方法是国内外诊断α地中海贫血基因缺失最常用的方法,较多实验室用Jf I跨越缺失基冈断裂点序列的Gap-PCR对缺失型α和β地中海贫血进行基因诊断 。
2.2.3 PCR-反向点杂交法(PCR-RDB)
原理是将一系列已知寡核苷酸探针固定于支持膜上,加入PCR产物进行液相探针杂交,可同时检测多种点突变。因该法快速、准确、所需样本量少,故已成为国内外实验室最常用的地中海贫血点突变检测方法,尤其适用于β地中海贫血点突变的检测 。但该法操作繁杂且易形成主观判断上的失误,故对实验室技术人员的技能要求较高,同时,如遇可疑结果须重复检测。
2.2.4 实时荧光PCR的探针溶解曲线分析(PM-CA)
原理是运用实时荧光检测技术结合探针溶解曲线分析技术,在异常扩增后检测PCR产物多色荧光溶解曲线从而鉴定多种突变 。我国学者严提珍等 分别采用PMCA方法与PCR-RDB方法对451份外周血标本和84份胎儿绒毛、羊水、脐带血产前诊断标本进行检测,并用DNA测序法进行验证,结果发现前者比后者有更高的基因诊断符合率,能够更准确地检测出多种基因突变类型。但该方法还处于完善阶段,对技术要求高,操作复杂且费用昂贵,故仅在少数几个大型临床实验室中开展,暂时未能在临床工作中大规模应用。
2.2.5 基因芯片
原理是通过微加工及微电子技术,在固相介质(如玻片、塑料、硅片、聚丙烯酰胺凝胶等)上固定序列已知的核苷酸探针,与已被荧光素或同位素标记的核酸序列进行杂交,可通过激光共聚焦显微镜上反应点的荧光强弱及位置,获得一组序列完整互补的探针序列,重组出靶核酸的序列 。其优点是高效、灵敏、快速,可短时间同时检测多种基因突变,但是成本昂贵,目前尚未在临床诊断中大规模应用。
2.2.6 DNA测序
方法是将标本DNA经PCR扩增后,在DNA自动测序仪上进行序列分析。常用于当其他基因检测法与临床表型不符合时的进一步验证,或者是分析未知基因突变时选用的检测方法。该方法是判断基因点突变类型及位置的金标准,但是该法对实验设备及标本DNA浓度要求较高并且所需时间较长,目前多在实验室中应用。
2.2.7 其他方法
其他基因检测方法较多,如等位基因特异性扩增技术(ARMS)适合针对某种小片段突变类型的特定检测;变性梯度凝胶电泳(DGGE)费用低廉,适合大规模的未知突变基因筛查;巢式PCR(又名套内PCR)是胚胎植入前常用的基因诊断方法;PCR限制性内切酶谱分析(PCR-SSCP)常用来检测未知基因突变,但影响结果的因素较多,不能用于大片段基因(超过400碱基)的检测。
2.3 胎儿产前诊断
目前胎儿产前诊断主要有胎儿基因诊断和超声检查。
2.3.1 胎儿基因诊断
胎儿基因诊断分为有创及无创性胎儿基因检查。何升等对1 072例高风险孕妇孕10~15周绒毛样本进行地贫基因检测,在产前诊断后1周内终止妊娠能有效避免重型地中海贫血患儿出生 。但通过穿刺手段采集绒毛、羊水等易对胎儿造成伤害,增加流产、宫内感染等风险。
1997年,Lo等 的研究发现妊娠母体血浆中存在胎儿的遗传物质,通过对胎儿遗传物质进行检测诊断能够避免有创检查带来的风险,更加安全。经过科学家们长时间的研究,无创性基因检测在2008年有了突破性进展,Galbiati等 利用PNA钳夹以微电子芯片技术检测孕妇血浆中游离的胎儿β珠蛋白基因突变,检测准确度为100%,特异性为98.3%。近年,对母体血浆中游离DNA的定量检测技术有了新的进展,利用母体血浆cff-DNA的二代测序技术,可对基因组范围的目标序列进行定量分析从而进行无创产前检测。我国开发了一种基于孕妇血浆DNA目标区域高深度测序,对可能携带致病性拷贝数变异(CNVs)的胎儿样本进行无创产前基因检测的方法,通过该方法,研究人员可以准确检测出胎儿是否患有地中海贫血 。相信在未来通过进一步的探索和完善,无创产前基因诊断能够更好地运用到临床工作中。
2.3.2 产前超声检查
产前超声检查具有安全、无创、简便、可重复等优点,报道显示其对地中海贫血组胎儿的正确诊断率可达92.3%。通过超声检测胎儿水肿成为重型地中海贫血产前筛查和诊断的常用方法,可作为基层医院地中海贫血产前诊断的选择。
----
我国β地中海贫血地区分布
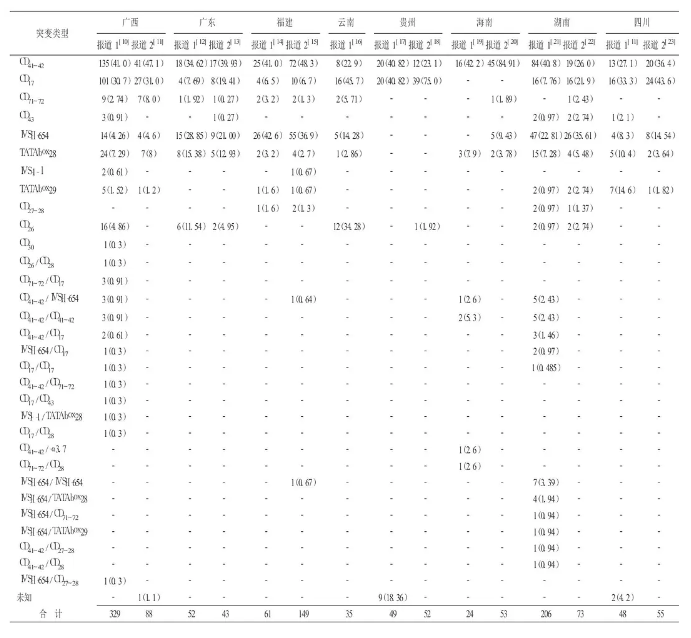
β地中海贫血是我国长江以南各省发病率最高、影响最大的遗传病之一, 尤以广西、广东、福建、云南、贵州、湖南等省最多。估计高发地区中2%~3%的人为该病基因携带者。大量调查表明, β地中海贫血的基因频率有地区差异, 而且不同的民族之间也有差异。
表1显示, 广西、广东、海南地区频率最高的都是CD41~42, 广西地区除了CD41~42, 按频率高低依次为CD17、TATAbox-28、CD26、IVS-Ⅱ-654、CD71~72;广东地区除了CD41~42, 按频率高低依次为IVS-Ⅱ-654、CD17、IVS-Ⅰ-1、CD43、CD71~72;海南地区除了CD41~42, 按频率高低依次为TATAbox-28、IVS-Ⅱ-654、CD71~72、CD17、CD26、CD1、TATAbox-28。福建地区频率最高的是IVS-Ⅱ-654, 其他基因按频率高低依次为CD41~42、CD17、CD71~72、TATAbox-28、TATAbox-29、CD27~28, 虽然最高频率的变异位点不是CD41~42, 但是和广西、广东、海南这三个地区有CD41~42、CD17、CD71~72、IVS-Ⅱ-654四个相同变异类型, 这4种突变类型在这4个地区占80%以上, 与徐湘民等报道相近, 但基因频率与报道的全国其他地区的频率稍有出入。广西、广东、海南和福建都处在中国的南端, 气候纬度接近, 同属于亚热带地区, 四省的地理位置和气候条件以及疟疾流行病相近存在很大的关系。流行病学、遗传学和体外实验研究结果表明, 人类β珠蛋白基因是受疟疾选择作用最明显的基因之一。流行病学研究结果显示, β珠蛋白基因常见的HbS(β6Glu※Val) 、HbC以及β地贫主要分布在非洲、地中海地区、中东、东南亚和中国南方等热带、亚热带地区, 与疟疾的流行区域基本一致。
中国不同省区β地中海贫血基因变异类型及频率分布
据报道β珠蛋白基因变异与人群的迁徙有着密切的关系(明末清初, 福建一部分人口迁入广东) , 广东地区的人群和福建地区有着很深的渊源, 且在迁徙过程中不仅发生生物遗传, 而且因各自地理位置、气候条件、疟疾的不同和人群相互融合、相互通婚产生了变化, 所以这两个地区的基因型既有相同点又有各自的特点。据Shih报道, 台湾常见的β珠蛋白基因变异类型为IVS-Ⅱ-654、CD17、TATAbox-28和CD41~42, 且IVS-Ⅱ-654最多见, 与福建地区报道相近, 推测与福建和台湾地区地理位置毗邻, 两地区人口之间相互迁徙交流有关。台湾地区因其存在本地特有的人群, 如高山族等, 而福建地区地理面积大而人群复杂, 所以两地区基因变异类型又有区别, 福建地区的基因变异类型多于台湾地区。
云南地区变异位点按频率高低依次为CD17、CD26、CD41~42、IVS-Ⅱ-654;贵州地区按频率高低分别是CD17、CD41~42、CD26, 云南、贵州两个地区频率最高的变异位点都为CD17, 有一定的相似性, 且都有CD17、CD41~42、CD26三个相同的变异位点。云南、贵州两个地区地理位置相毗邻, 都处于中国的西南云贵高原, 气候纬度接近, 群体基因的选择压力接近;这两个地区处于边远、交通比较闭塞, 族内婚配、家族发病聚集性和通婚地域半径狭小, 基因频率高低和顺序又有各自不同的特点。
