下一代癌症靶向药物的纵览与展望
外科手术、化疗、放疗是癌症的传统三大治疗手段,这些疗法在过去和现在为治疗癌症发挥了重要作用,但其经常存在不够精准,存在“伤敌一千,自损八百”的情况。随着分子生物学的发展和人们对人体生命过程理解的加深,癌症出现了第四大治疗手段——靶向药物。
通过研究发现癌症分子病理过程的关键调控分子,然后在药物或其载体中包含与这些靶标(target)特异性结合的配体结构,使药物能够精确地与靶标发生作用,从而杀伤或抑制癌细胞,这就是癌症靶向药物的基本原理。相对于癌症传统三大疗法,靶向药物具有针对性强、副作用小的优势,符合“精准医疗”的理念,已经成为癌症治疗的主流方法。2017年FDA批准的14种抗癌新药全部属于靶向药物。
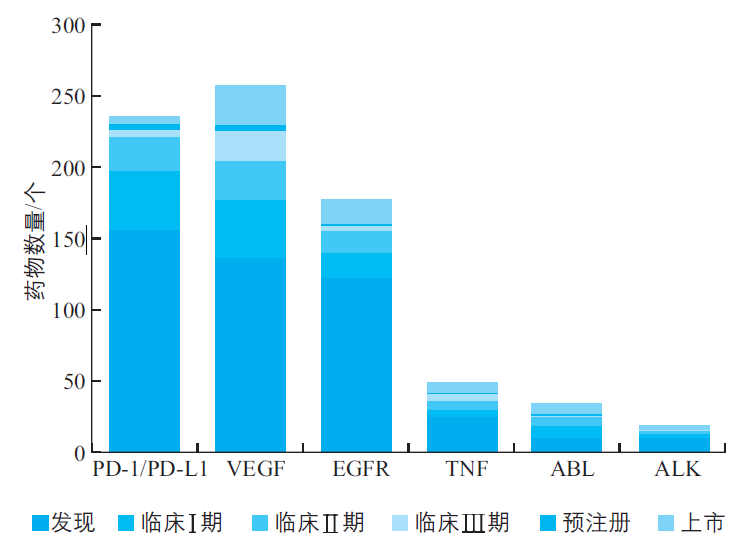
据统计,全球癌症靶向药物主要集中于一些认知相对充分的靶点,如PD-1/PD-L1、VEGF、EGFR、Bcr-Abl、ALK等。根据数据库Cortellis在2018年的数据,针对PD-(L)1、VEGF和EGFR这3个靶点的在研和上市药物占癌症靶向药物总量的86.8%。过度同质化竞争、耐药性的进展、患者的临床需求,推动了制药企业和研发机构纷纷加入下一代癌症靶向药物的研发竞赛。
一、下一代激酶抑制剂
据统计,美国FDA已经批准了50多款激酶抑制剂,还有更多的正待审查。此外,至少有20种激酶通路已经被验证,包括BTK、VEGFR等,但是新的激酶抑制剂的研发竞赛也在火热进行中。
随着对激酶抑制剂的研发不断加速,投资规模持续增长,对于靶点的选择也越来越精确。以Kronos Bio收购Gilead的SYK抑制剂为例,这些药物是为急性髓系细胞白血病(AML)患者开发的,并且这些患者的转录因子HOXA9/MEIS1过表达,或者存在FLT3基因变异。
SYK抑制剂
2020年7月,Kronos Bio投资1.2亿美元购买Gilead的SYK抑制剂产品组合,这表明尽管细胞基因疗法正在兴起,酪氨酸激酶阻滞剂仍然是肿瘤学研究中的热门。这个组合包括临床阶段化合物entospletinib(GS-9973),已在肿瘤患者的I期和II期临床试验中进行了评估,以及lanraplenib,已在自身免疫性疾病患者的II期临床试验中得到了评估。
SYK为脾脏酪氨酸激酶(spleen tyrosine kinase),是全球创新药研发企业靶向的几种新兴酪氨酸激酶之一。SYK被认为是多种癌症的促进因子,能够促进酪氨酸上的蛋白磷酸化,并参与B细胞抗原受体(BCR)信号通路,是多种亚型B细胞淋巴瘤和自身免疫性疾病的既定靶点。目前有几种药物可以有效抑制SYK,但是脱靶效应是主要限制因素。
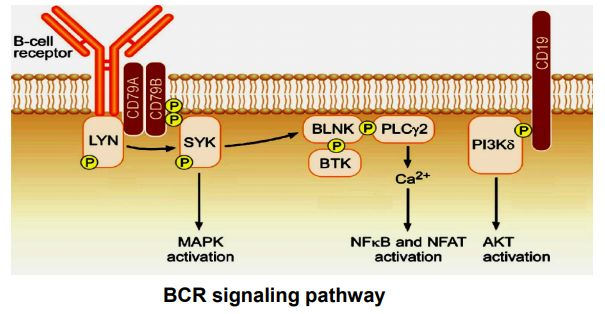
2018年,和黄医药公布了在中国开展的一项评估SYK抑制剂HMPL-523治疗复发或难治性B细胞淋巴瘤患者的I期试验初步结果,以及在澳大利亚开展的一项健康志愿者I期剂量递增研究结果,均表明总体耐受性良好。该药针对广泛的血液学癌症,继续在澳大利亚和中国进行Ib期剂量扩充试验。和黄医药还在2019年8月启动了一项I期临床试验,评估该药用于免疫性血小板减少症(ITP)患者的治疗。
KRAS抑制剂
RAS(鼠肉瘤病毒)基因是人类癌症中最常见的突变基因,在90%的胰腺癌、45%的结肠癌和35%的肺癌中存在突变。KRAS则是RAS基因家族中最常发生突变的亚型,在RAS基因突变中占比高达86%。KRAS蛋白的突变形式包括KRAS-G12突变(密码子-12的甘氨酸变异为半胱氨酸)和KRAS-G12D突变(密码子-12的甘氨酸变异为天冬氨酸)。KRAS-G12突变在NSCLC中(45-50%)占主导地位,KRAS-G12D突变在胰腺癌(61%)、结肠癌(42%)中占主导地位。
从机制上说,靶向KRAS的药物具有广阔的市场前景,然而RAS近乎球形的分子空间结构使其缺乏药物结合位点。安进首先实现了突破,sotorasib(AMG510)可以与KRAS-G12C蛋白共价结合,将其不可逆地锁定在失活状态。传统意义上不可成药靶点的突破引发了制药行业和投资界的浓厚兴趣,使KRAS成为近两年最热门的靶点之一。2019年9月,早期数据显示,10名非小细胞肺癌(NSCLC)患者在最高剂量的试验中对AMG510治疗都有响应。安进在2020年5月的ASCO(美国临床肿瘤学会)会议上报告了该药在其他几种肿瘤中的试验数据。然而,业界对此回应不温不火,分析认为NSCLC是AMG510的最佳前景。
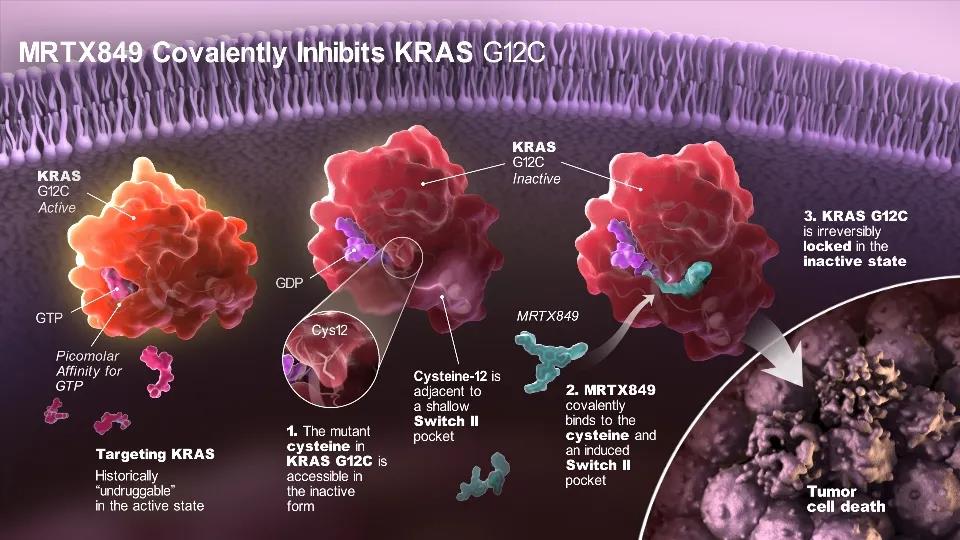
与此同时,安进还面临着来自Mirati的竞争。2020年10月,Mirati公布了KRAS-G12C变异亚型抑制剂adagrasib(MRTX849)在针对NSCLC的I期和II期临床试验产生45%的应答率,比安进的sotorasib在类似试验的35%略高。然而,Mirati是基于可分析人群得到的客观缓解率(ORR),而安进是基于所有参加试验人群,如果用安进的方式进行计算,adagrasib的ORR为33%,甚至略低于sotorasib。Adagrasib在KRAS-G12C突变的结直肠癌患者中的应答率低至17%,但比sotorasib的7%略高。总体来说,根据现有数据,Mirati的adagrasib略胜一筹。
勃林格殷格翰(Boehringer Ingelheim)、Revolution Medicine和美国默克(Merck & Co)都希望加入KRAS抑制剂研发竞赛。美国默克出资25亿美元,以参与大鹏制药(Taiho Pharma)和Astex包含KRAS抑制剂的管线。
靶向KRAS有一定的风险。随着这些抑制剂进入临床试验阶段,投资者无疑会关注其安全性。今年7月,礼来(Eli Lilly)表示,在I期试验中发现了意想不到的毒性后,其放弃了KRAS的研发计划。
AKT抑制剂
在酪氨酸激酶抑制剂研发领域的另一个新靶点是蛋白激酶B(AKT),它参与调节雌激素受体(ER)。阿斯利康(AstraZeneca)正在测试其AKT抑制剂capivasertib联合氟维司群对ER+乳腺癌的疗效。2019年夏,II期临床试验数据显示,与氟维司群单药相比,联合用药使疾病进展时间延长了一倍,延长了患者6个月的生命。
由于面临乙肝的管线变更带来的一些挑战,罗氏放弃了将AKT抑制剂ipatasertib(RG7440)开发为三阴性乳腺癌(TNBC)患者的新辅助治疗,但是对AKT抑制剂仍然持乐观态度。它正在招募患者进行ipatasertib针对其他适应证的试验,包括乳腺癌、卵巢癌、前列腺癌,以及难以治疗的脑癌胶质母细胞瘤。
2020年6月,罗氏公布的III期临床试验数据显示,ipatasertib与醋酸阿比特龙联用改善了患有PTEN(10号染色体上磷酸酶及张力蛋白同源基因)缺失异常的前列腺癌患者的无进展生存(PFS)。虽然该药未能在更广泛的人群中改善疗效,但鉴于总体生存数据仍在不断成熟,罗氏认为这些发现仍然令人鼓舞,因为它有机会从阿斯利康capivasertib的目标市场中分一杯羹。阿斯利康的AKT抑制剂capivasetib正在进行两项III期乳腺癌试验,但在前列腺癌的试验中尚未超过I期。
然而,AKT药物的研发并不十分理想,GSK出于安全性考虑终止了GSK690693的研发,而默克(Merck & Co)的MK-2206疗效不佳。上述AKT抑制剂的研发失败使阿斯利康的capivasertib和罗氏的ipatasertib成为该领域的领先者,
MET抑制剂
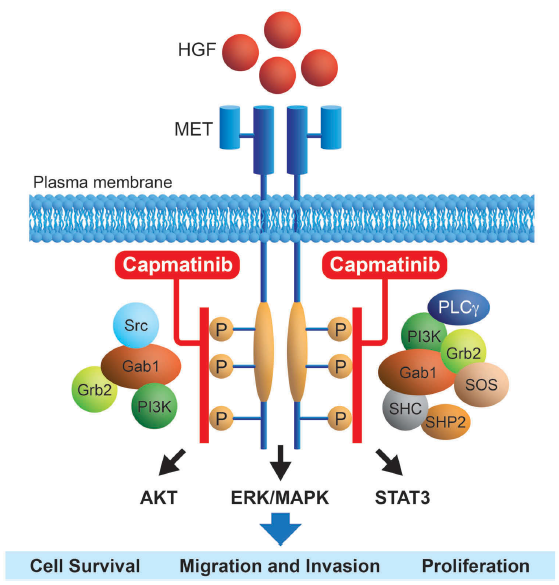
MET是另一个新兴靶点,但也面临着一些挑战。MET也被称为cMET,是一种酪氨酸激酶,在几种癌症类型中过度表达或突变,帮助促进肿瘤细胞的存活和复制。
2019年10月,阿斯利康放弃了其用于治疗乳头状肾细胞癌的MET抑制剂savolitinib的研发,因为该药在III期试验中的疗效未能超越辉瑞的多受体酪氨酸激酶(RTK)抑制剂苹果酸舒尼替尼。阿斯利康目前正在对一组肺癌患者进行savolitinib的单独测试,这些患者在接受了EGFR抑制剂奥希替尼治疗后癌症进展或对酪氨酸激酶抑制剂产生耐药性。
诺华和德国默克(Merck KGaA)也参与了MET抑制剂的研发竞赛。诺华的坚持首先获得了回报,2020年5月美国FDA批准了Tabrecta(capmatinib)用于治疗MET外显子14跳跃突变的NSCLC患者。此项批准是基于一项试验,试验显示该药对初治患者的响应率为68%。与此同时,默克的tepotinib对同一组NSCLC患者的总有效率为45%至50%,并且已经被美国FDA授予快速通道资格。
二、针对确证靶点的耐药性
癌症治疗领域面临的最大挑战是,患者在接受靶向疗法之后,往往会对治疗产生耐药性。这就是为何该领域的下一个前沿是识别产生耐药突变的原因和模式,并找到针对性的新药。
溶剂前沿突变
在使用酪氨酸激酶抑制剂的患者中,最常见的突变被称为“溶剂前沿”突变。溶剂前沿是激酶表面许多抑制剂附着的区域,这一机制被耐药突变破坏。
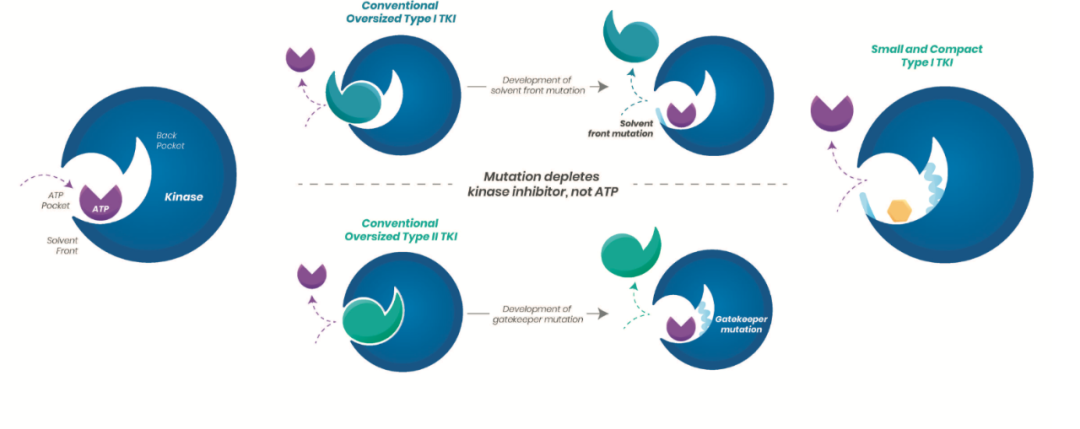
Turning Point Therapeutics公司的repotrectinib(TPX-0005),是一种低分子量、大环酪氨酸激酶抑制剂,靶向ROS1、TRK和ALK。该药可有效地与活性激酶构象结合,避免来自各种耐药突变的空间干扰。紧凑而刚性的三维结构使repotrectinib能够精确且有效地深入到激酶的ATP结合口袋中,从而抵抗激酶的溶剂前沿和gatekeeper突变。在一项针对NSCLC的II期临床试验的中期分析中,该药的客观响应率为86%,这使得该公司可能会通过获得FDA的加速审评审批程序快速上市。
抗HER2耐药
目前在激酶抑制方面的大部分研发工作都是针对已知的癌症通路,但目标是做的更好。例如,Seagen(原Seattle Genetics)正在带头改进HER2乳腺癌的治疗。2018年1月,Seattle Genetics以69%的溢价收购了Cascadian Therapeutics及其靶向HER2的酪氨酸激酶抑制剂tucatinib(图卡替尼),该药针对罗氏的HER2三剑客——Herceptin、Perjeta或Kadcyla停止响应的乳腺癌患者开发。
试验数据显示,在赫赛汀和卡培他滨的基础上增添tucatinib,比仅使用赫赛汀和卡培他滨降低了46%的疾病进展或死亡风险,并且显著延长了总生存期,这令投资者们大为赞叹。2019年12月,美国FDA授予tucatinib突破性疗法认定,与曲妥珠单抗和卡培他滨联用,治疗不可切除的局部晚期或转移性HER2阳性乳腺癌患者。2020年4月,Tukysa(tucatinib)获得了美国FDA的批准,适应证同上。
RET融合耐药
耐药性的另一罪魁祸首是RET融合,一些EGFR阳性肿瘤会随着时间的推移而发展出这种基因异常。2020年5月,礼来(EliLilly)的RET抑制剂selpercatinib提前获得了美国FDA的批准,用于治疗有RET融合的甲状腺癌。该药是2019年礼来以80亿美元收购Loxo Oncology时获得的资产之一。
罗氏和礼来在RET药物研发上势均力敌。罗氏于2020年7月斥资7.75亿美元获得了BlueprintMedicines的pralsetinib的专利权,该药对NSCLC的有效率为65%,对甲状腺癌的有效率则高达91%。2020年9月,Gavreto(pralsetinib)获得FDA批准,用于治疗RET融合阳性的NSCLC。
三、下一代免疫肿瘤学靶点
第一波免疫肿瘤学药物针对免疫检查点(immune checkpoints),产生了针对PD-(L)1的重磅药物。下一波免疫肿瘤学药物包括第二代检查点抑制剂,以及能与多种新出现的免疫靶点相互作用的药物。目前的研究正在努力了解肿瘤微环境,如果免疫靶点与杀伤T细胞相互作用的机制能够被充分理解,那么未来有望成功开发出更多通过调节肿瘤微环境来治疗癌症的有效疗法。
第二代免疫检查点TIGIT
一个引起免疫肿瘤学界注意的新兴免疫靶点是TIGIT,它是指带有Ig和ITIM结构域的T细胞免疫受体,存在于某些T细胞和自然杀伤细胞上。TIGIT可以通过多种机制抑制T细胞和自然杀伤(NK)细胞。TIGIT与树突状细胞上的CD155相互作用,通过脂多糖刺激增加IL-10分泌水平,降低IL-12分泌水平,并且抑制体内T细胞活化。TIGIT对自然杀伤细胞(NK)的抑制作用可被阻止其与CD155相互作用的抗体阻断。
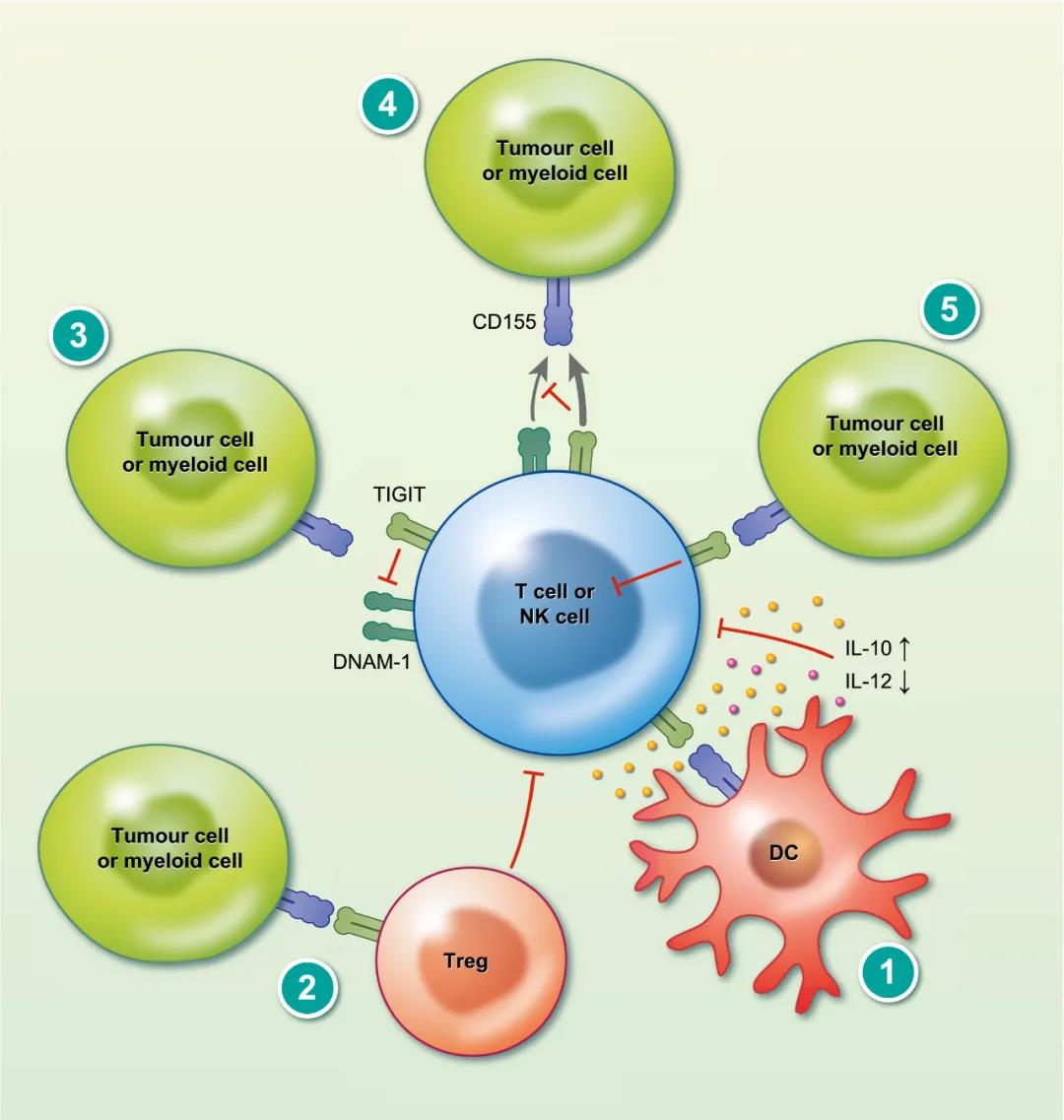
2018年,美国默克报告称,在一项进行中的I期试验中,其在研TIGIT靶向药物MK-7684与PD-1抑制剂Keytruda联用治疗晚期实体瘤,使40%的患者病情稳定。默克正在招募患者进行NSCLC联合治疗的II期试验。默克在2016年底就已经开始MK-7684的人体试验,目前正准备启动一系列亚研究,以评估该药物与Keytruda联用在黑色素瘤患者中的疗效。
紧随其后,罗氏推出了TIGIT组合策略。2020年5月,ASCO年会上,罗氏旗下的基因泰克(Genentech)报告其抗TIGIT候选单抗tiragolumab联合PD-L1阻断剂Tecentriq(atezolizumab,阿特珠单抗),可使31%的NSCLC患者的肿瘤缩小,轻松超越Tecentriq单药疗效。罗氏有8项关于tiragolumab的临床试验已经或即将开始招募患者,其中包括两项肺癌III期研究。
吉利德不甘落后,斥资近20亿美元与Arcus达成交易,以获得后者的抗TIGIT候选药物AB154的部分权益。Arcus已经将AB154推进到II期临床试验,联合AB154与自身在研的PD-1抑制剂zimberelimab治疗NSCLC。Arcus希望能够证明AB154可以提高PD-1抑制剂的疗效。目前正在研发TIGIT药物的生物科技公司还有iTeos Therapeutics和Mereo BioPharma,这两者可能成为通过并购进入TIGIT领域的龙头企业的潜在标的。
众多公司和投资者都在TIGIT上押注,将其视为长期寻求的能够将PD-1/PD-L1抑制剂的疗效提升至新水平的靶标。然而,不要忘记增强检查点抑制剂疗效的早期努力曾经完全熄火,其中最富戏剧性的是Incyte公司的epacadostat的失败。该药通过抑制IDO1(吲哚胺2,3-双加氧酶-1)来激活免疫系统,早期小型临床试验数据良好,被认为极有前景。然而,在2018年4月,Incyte与默沙东大规模联用epacadostat和Keytruda的III期临床试验遭遇失败。随后,其他公司宣布停止各自正在研发的IDO抑制剂抗癌药项目。
制药行业在IDO靶点上始于用力过猛,终于惨淡收场。究其原因,业界一方面对于IDO的作用机制并没有理解透彻,另一方面急于找到一个能够扩大抗PD-1单抗适用人群的方法。在急于求成的心理驱使下,研发节奏过于激进,导致了在大规模试验中的失败。
CD47靶向药物的迭代
另一个免疫靶点CD47长期以来一直备受关注,这是一种广泛表达于多种癌细胞表面的糖蛋白,能够与肿瘤吞噬细胞表面的SIRP&α;结合释放“别吃我”的信号,阻止巨噬细胞的吞噬作用。阻断这一信号通路可以使巨噬细胞攻击肿瘤细胞,使CD47成为有开发前景的肿瘤免疫靶点之一。然而,由于大多数CD47抗体都会与红细胞结合,在临床上可能会引起溶血性贫血,这迫使许多企业不得不停止开发CD47抗体。新一代CD47抗体在清除肿瘤细胞的同时,要尽量减少与红细胞的结合。
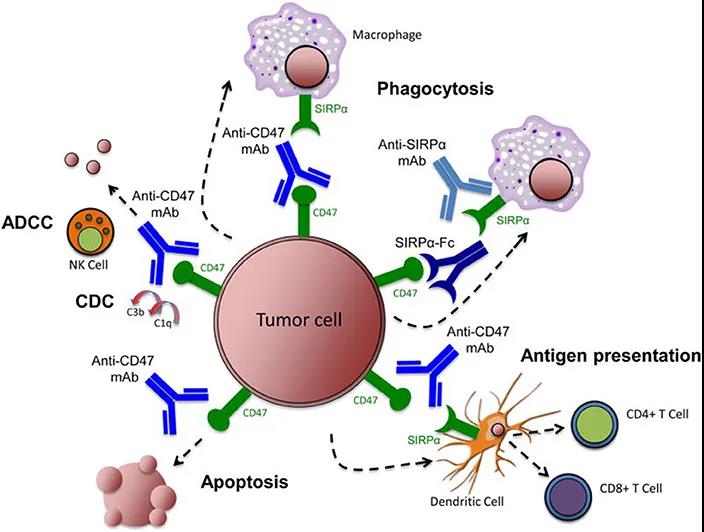
2020年3月,吉利德在CD47领域投下了迄今为止最大的赌注,斥资49亿美元收购了Forty Seven和中期在研药物magrolimab(5F9)。2020年6月,Forty Seven在欧洲血液学协会大会上公布了Ib期临床试验数据。Magrolimab与阿扎胞苷联用,在既往未接受治疗的高风险骨髓增生异常综合症(MDS)的患者中达到91%的客观缓解率(ORR)和42%的完全缓解率(CR),在初治急性髓系白血病(AML)患者,达到64%的ORR和56%的CR。2020年9月,magrolimab被FDA授予突破性疗法认定,适应症为新确诊的MDS。
2020年9月,艾伯维加入了开发抗CD47药物的竞赛,出资20亿美元与天境生物合作研发lemzoparilimab(TJC4),创下了中国创新药企对外授权转让的新高。多项临床前研究充分证明了该药的血液学安全优势和突出的抗肿瘤活性。在癌症患者中的I期临床试验进一步验证了与其他CD47靶向药物的差异化。天境生物将继续在美国推进lemzoparilimab与Keytruda联合治疗实体瘤,以及与利妥昔单抗联合治疗淋巴瘤的临床试验。
围绕PD-1靶点的改进
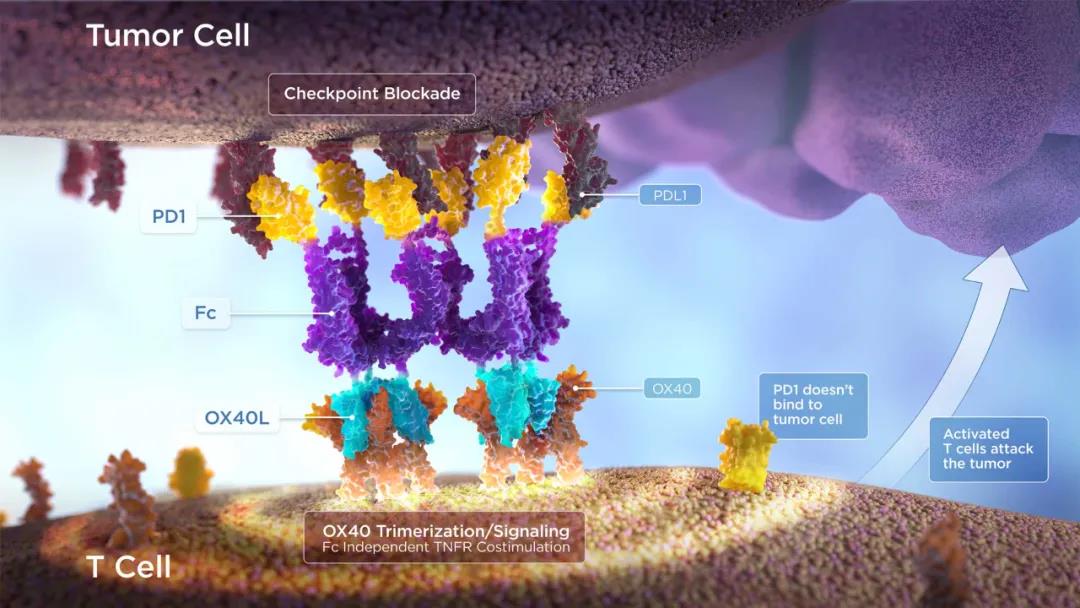
免疫肿瘤学的大部分研发工作集中在改善疗法对已得到确认的免疫检查点的靶向性,如PD-1。例如,初创公司Shattuck Labs正在基于自有ARC(激动剂重定向检查点)平台,研发一类双功能融合蛋白,将免疫检查点受体(PD-1,SIRP&α;,TIGIT)与肿瘤坏死因子(TNF)配体(OX40L,41BBL,CD40L,LIGHT)通过不活跃的Fc域连接。这种设计与基于抗体的疗法相比,具有两个明显优势:在没有Fc介导的交联的情况下激活肿瘤坏死因子超家族(TNFRSF),并且在单一化合物中结合了检查点封锁和免疫共刺激。
SL-279252(PD1-Fc-OX40L)含有PD1的胞外域结构,通过IgG4 Fc连接蛋白与OX40配体(OX40L)的胞外域结构连接,并且OX40L侧的适当折叠形成三聚体OX40L,与OX40结合时可以有效地刺激免疫细胞的激活途径。该药能够与肿瘤细胞上的PD-L1和T细胞上的OX40结合,解除PD-1/PD-L1信号通路对T细胞的免疫抑制,同时激活OX40活化通路,导致肿瘤微环境中的T细胞被激活。简言之,该药不仅可以消除免疫系统的刹车,还可以促进T细胞攻击癌细胞。
目前,免疫检查点CTLA-4的靶向药物市场由BMS的Yervoy(ipilimumab,伊匹单抗)垄断,该药在2019年的全球销售额约为15亿美元。现在有一些公司希望为抗CTLA-4药物市场提供一些新选择,如同时靶向PD-1和CTLA-4的双特异性抗体。MacroGenics正在研发一种DART模式的四价双特异性抗体MGD019,已于2018年12月开始了针对不可切除/转移性癌症的临床I期剂量递增试验。中山康方的AK104为四价双特异性抗体,基于自有的TETRABODY技术设计,避免Fc引起的副作用,能够与肿瘤浸润淋巴细胞(TILs)优先结合,而非正常外周组织淋巴细胞。针对宫颈癌的2/3线治疗,AK104已经在中美两地进入临床II期关键试验。
四、结语
在与癌症漫长的抗争历史中,人类长期处于劣势。人类面对癌症,甚至可以说是小米加步枪对飞机与大炮。现有的癌症靶向药物基本都经历了漫长的研发与不断的试错的过程,都是来之不易的成果。面向未来,我们一方面需要加深对现有靶点作用机制的认知,对现有癌症靶向药物进行迭代,将与癌症战斗的刀枪打磨地更加锋利。另一方面,则在生命科学上不断开拓,随着对癌症病理和人体生命过程的理解不断深入,未来必定会发现更多有潜力的靶点与更多更好的靶向药物。实现这一目标需要科学界、制药界和投资界的通力合作。
