去磷酸化抑制 CHEK1 可破坏 S 期限制点并诱导 CML 细胞凋亡
去磷酸化抑制CHEK1可破坏S期限制点并诱导CML细胞凋亡
王 方1,王 昕2,王亚文1, 2
Dephosphorylatory inhibition of CHEK1 abrogates S phase checkpoint and induces apoptosis of CML cells
WANG Fang1, WANG Xin2, WANG Yawen1,2
作者单位
1. 西安交通大学第一附属医院检验科,陕西 西安 710061;2. 西安交通大学第一附属医院生物样本信息资源中心,陕西 西安 710061
基金项目
国家自然科学基金资助项目(编号:81600134)
AUTHORS FROM
1. Department of Laboratory Medicine, First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, Shaanxi Province, China; 2. Biobank, First Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710061, Shaanxi Province, China
GRANT
National Nature Science Foundation of China(No. 81600134)
[摘要]
目的:探讨细胞周期检测点激酶1(checkpoint kinase 1,CHEK1)参与拮抗慢性粒细胞白血病(chronic myelogenous leukemia,CML)细胞凋亡的分子机制。
方法:通过GEO(Gene Expression Omnibus )数据库内置的R语言程序进行CML细胞的基因表达数据分析,选取差异表达的靶基因。采用实时荧光定量PCR法及蛋白质印迹法检测8例CML患者外周血中CHEK1 mRNA和蛋白的表达水平,并分析其与CML病情进展的关系。用针对CHEK1的抑制剂SCH900776抑制CHEK1磷酸化后,采用FCM法检测K562和Ku812细胞周期及凋亡的变化,并用蛋白质印迹法检测下游相关分子的表达水平变化。
结果:通过对GEO数据库中3项CML基因表达数据进行分析,得到共同的差异表达基因共21个,其中12个为表达上调,9个为表达下调;选取CHEK1作为研究对象。CML患者外周血中CHEK1 mRNA和蛋白表达水平均明显高于健康体检者(P值均<0.05),且急变期CHEK1 mRNA表达水平明显高于慢性期(P<0.05)。CHEK1及其磷酸化蛋白的表达水平随病情进展亦有增高的趋势,且在伊马替尼处理后未有明显变化。抑制CHEK1磷酸化可激活半胱天冬酶-3,并显著诱导K562和Ku812细胞凋亡(P 值均<0.01)。抑制CHEK1磷酸化后,S期细胞减少,G2/M期细胞增多,S期相关蛋白细胞周期蛋白E(cyclin E)表达水平下调,而DNA损伤标志物&γ;-H2XA 表达水平升高,同时p53家族成员中p53和p73的表达均明显上调(P 值均<0.05)。
结论:CHEK1去磷酸化失活可逆转细胞S期阻滞,破环DNA损伤修复,进而导致P73升高,诱导细胞凋亡。
[关键词]白血病,髓系,慢性,Bcr/Abl阳性;S期细胞周期检查点;DNA损伤;细胞凋亡
[ABSTRACT]
Objective:To explore the molecular mechanism of checkpoint kinase 1(CHEK1) mediating apoptosis resistance of chronic myelogenous leukemia(CML) cells.
Methods: CHEK1 was selected as the target by analyzing 3 gene expression data studies of CML cells in Gene Expression Omnibus(GEO) database using the inner R program. The expression of CHEK1 mRNA and protein in peripheral blood of 8 patients with CML was detected by real-time fluorescent quantitative PCR and Western blotting, and the relationship between CHEK1 expression and the progression of CML was analyzed. After phosphorylation of CHEK1 was inhibited by CHEK1 inhibitor SCH900776, the cell cycle and apoptosis of K562 and Ku812 cells were detected by FCM, and the expressions of downstream related molecules were detected by Western blotting.
Results: Out of 21 genes that differentially expressed in all 3 studies of GEO database(9 down and 12 up), CHEK1 was selected as an upregulated gene for the further research. Both CHEK1 mRNA and protein levels were increased in CML patients as compared with the healthy controls. Besides, the CHEK1 protein and phosphorylated CHEK1 were further upregulated in CML patients at blast crisis stage(P < 0.05), which were not affected by imatinib treatment. Inhibition of CHEK1 phosphorylation significantly triggered the apoptosis of K562 cells (P < 0.01) and Ku812 cells(P < 0.001) through 半胱天冬酶-3 activation. Dephosphorylation of CHEK1 attenuated the expression of S-phase related protein cyclin E, resulting to the raise of G2/M phase and the failure of DNA damage repair which was reflected by up-regulating &γ;-H2XA level. In addition, the expressions of two P53 family members P53 and P73 were obviously enhanced after CHEK1 dephosphorylation(P < 0.05).
Conclusion: Dephosphorylation of CHEK1 can reverse S phase arrest and destroy DNA damage repair, leading to increase of P73 level and induction of apoptosis.
[Key words]Leukemia, myelogenous, chronic, Bcr/Abl positive; S phase cell cycle checkpoints; DNA damage; Apoptosis
慢性粒细胞白血病(chronic myelogenous leukemia,CML)是起源于造血干细胞的恶性增殖性疾病。t(9,22)染色体易位形成的BCR/ABL融合基因是CML的分子标志,该基因编码非受体型酪氨酸激酶BCR/ABL,通过自身磷酸化活化,激活下游信号转导和转录活化因子5(signal transducer and activator of transcription 5,STAT5)/Bcl-2、磷酸肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)/蛋白激酶B(protein kinase B,PKB,又称为AKT)及Ras/丝裂原细胞外激酶(mitogen extracellular kinase,MEK)等多条信号通路,调节细胞增殖及凋亡[1]。针对Bcr/Ab激酶所开发的酪氨酸激酶抑制剂(tyrosine kinase inhibitors,TKIs)能够特异性抑制BCR/ABL激酶活性,进而阻断下游信号。
作为首个靶向药物,伊马替尼(imatinib)自1991年被美国食品和药物管理局(Food and Drug Admistraton,FDA)批准上市以来,为CML治疗带来了革命性的突破,极大程度改善了患者的预后[2]。随后的研究发现,BCR/ABL基因T315I点突变及BCR/ABL基因过表达都能够导致伊马替尼耐药[3-5],尤其是进展期的患者。为了克服这一缺陷,二代TKIs如Dasatinib和Nilotinib等被陆续开发并应用于临床。二代TKIs能有效逆转伊马替尼耐药[6],但仍存在一部分患者具有原发耐药(primary resistance)特征,这类患者在初始使用TKIs时便疗效欠佳。有研究发现,CML原发性耐药在慢性期、加速期及急变期的发生率分别为5%、24%及66%[7]。此外,即使敏感患者在初次获得缓解后,仍有20%~30%患者可能发
生复发。因此,TKIs虽能有效改善病情,但距离CML治愈仍有大量问题需要解决[8]。寻找替代治疗靶点是治愈CML亟待解决的问题之一。
本研究通过对基因表达谱分析发现,细胞周期检测点激酶1(checkpoint kinase 1,CHEK1)在CML细胞中表达显著升高,且不依赖于BCR/ABL激酶活性;而抑制CHEK1活性对肿瘤细胞具有明显的促凋亡效应,提示CHEK1作为一个潜在的治疗靶点值得进一步研究。
1 材料与方法
1.1
主要试剂
CHEK1抑制剂SCH900776和Bcr/Abl抑制剂伊马替尼均购自美国Selleck公司。细胞凋亡及周期检测试剂框购自杭州联科生物技术股份有限公司。外周血单个核细胞分离液购自天津灏洋生物制品科技有限公司。TRIzol试剂框购自美国Invitrogen公司。实时荧光定量PCR试剂框购自美国Bio-Rad公司。兔抗人磷酸化细胞分裂周期素25A(phospho-cell division cycle 25A,p-CDC25A)、CHEK1、磷酸化CHEK1(Ser345)[phospho-CHEK1(Ser345),p-CHEK1(Ser345)]、半胱天冬酶-3、磷酸化细胞周期蛋白依赖性激酶2(phospho-cyclin-dependent kinase 2,p-CDK2)和&γ;-H2AX单克隆抗体均购自美国Cell Signaling Technology公司。兔抗人GAPDH单克隆抗体购自杭州贤至生物科技有限公司。鼠抗人p53、p63和p73单克隆抗体及辣根过氧化物酶(horseradish peroxidase,HRP)标记的羊抗鼠或兔IgG购自美国Santa Cruz公司。
1.2
细胞培养
CML细胞系K562和Ku812均购自北京北纳创联生物技术研究院,用含10%胎牛血清的RPMI 1640培养液,置于37 ℃、CO2体积分数为5%的培养箱中培养,收集对数生长期细胞用于后续实验。细胞处理分3组:未处理组,仅加入对照溶剂ddH2O;伊马替尼组,加入终浓度为1 μmol/L伊马替尼;SCH900776组,加入终浓度为500 nmol/L SCH900776。药物处理48 h后收集细胞,采用蛋白质印迹法检测相关蛋白的表达水平,以及FCM法检测细胞周期及凋亡情况。
1.3
临床样本处理
筛选西安交通大学第一附属医院2019年1月—2019年6月确诊的CML患者8例,其中慢性期5例,急变期3例;均为初诊患者,平均年龄48岁。选取同期年龄相近的正常体检者4例,作为对照。收集外周血标本,并根据人外周血单个核细胞分离试剂说明书,采用密度梯度离心法收集单个核细胞,然后立即用于RNA及蛋白提取,或于含10%胎牛血清的RPMI 1640培养液中短暂培养。
1.4
荧光定量PCR法检测CHEK1表达
用TRIzol试剂提取上述外周血单个核细胞中RNA,用Mini Drop微量分光光度计检测A260和A280,计算RNA总量。荧光定量PCR体系:SYBR green mix(2×)10 μL、iScript反转录酶0.25 μL、上下游引物各0.5 μL、RNA 500 ng,然后用ddH2O补足体积至20 μL。反应条件:50 ℃反转录10 min;95 ℃预变性1 min;95 ℃ 15 s,60 ℃ 30 s,45个循环;采用2-&δ;&δ;Ct法进行相对定量分析。CHEK1基因引物:上游引物序列为5’-TGCGTTGTAAGATTTATTTTGGCT-3’,下游引物序列为5’-AGTTTCCCGGAGAAAGCGAG-3’;内参&β;-actin基因引物:上游引物序列为5’-CTCCATCCTGGCCTCGCTGT-3’,下游引物序列为5’-GCTGTCACCTTCACCGTTCC-3’。
1.5
蛋白质印迹法检测CHEK1及相关蛋白表达
收集外周血单个核细胞或抑制剂处理后的K562和Ku812细胞106个/100μL,采用含1%苯甲基磺酰氟的RIPA裂解液于冰上裂解30 min,提取总蛋白。通过BCA法测定蛋白浓度,然后采用SDS-PAGE法分离蛋白质。电泳条件:浓缩胶和分离胶中聚丙烯酰胺密度分别为6%和10%~12%,蛋白上样量为20~50 μg,120 V恒压电泳2 h。电泳结束后,以恒流湿转法转膜,40 mA转6~8 h,然后将PVDF膜封闭于含5%的脱脂奶粉的封闭液中,室温处理1 h。分别加入工作液体积稀释比例为1∶1 000的兔抗人CHEK1、p-CHEK1(Ser345)、半胱天冬酶-3、p-CDC25A、p-CDK2、&γ;-H2AX和GAPDH单克隆抗体,以及鼠抗p53、p63和p73单克隆抗体,4 ℃孵育过夜,然后加入HRP标记的羊抗鼠/兔IgG(二抗),室温温育1 h,采用化学发光法检测蛋白表达信号。
1.6
FCM法检测细胞凋亡和细胞周期
收集抑制剂处理后的K562和Ku812细胞,用预冷的PBS洗3次后,加入结合缓冲液重悬细胞,调整细胞密度为106个/mL。取100 μL细胞悬液,加入藻红蛋白(phycoerythrin,PE)-AnnexinⅤ和7-氨基-放线菌素D(7-amino-actinomycin D,7-AAD)染料各5 μL,室温避光反应30 min后,使用流式细胞仪检测细胞凋亡率。细胞周期检测时,细胞需在收集后用70%乙醇溶液4 ℃固定30 min,然后用预冷的PBS洗涤2次;取100 μL细胞悬液加入20 μL PI染液,室温避光染色60 min;上流式细胞仪检测细胞周期分布情况。
1.7
统计学方法
应用SPSS 20.0软件进行统计学分析。本研究中各项实验均孤立重复3次,资料以 ¡À s表示;多组间比较采用方差分析,组内两两比较采用LSD-t法检验。以P<0.05认为差异有统计学意义。
2 结 果
2.1
数据库中差异表达基因分析
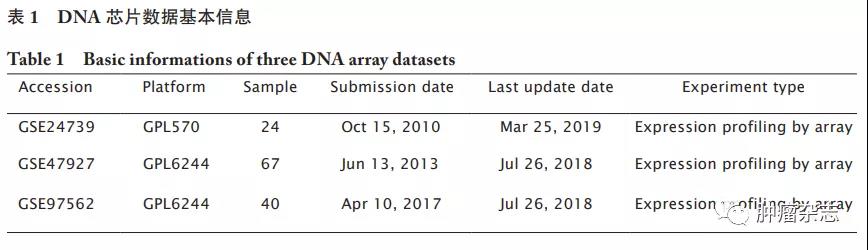
选取GEO(Gene Expression Omnibus)数据库(https://www.ncbi.nlm.nih.gov/geo)中3项关于CML基因表达的芯片数据进行差异表达基因分析,基本信息如表1所列。数据分析采用内置R程序(Analyze with GEO2R),将差异倍数(fold change,FC)按照|log2FC|>1且P<0.05的条件筛选,得到各项研究的差异表达基因。如图1所示:GSE24739、GSE47927和GSE97562数据集分别得到96、113和214个下调基因(图1A),以及150、273和175个上调基因(图1B)。其中,9个下调基因和11个上调基因(图1C)为3项研究所共有,基因名称如图1C中所示,后续选取上调表达基因CHEK1作为进一步的验证和研究对象。
2.2
CML患者外周血中CHEK1基因表达水平
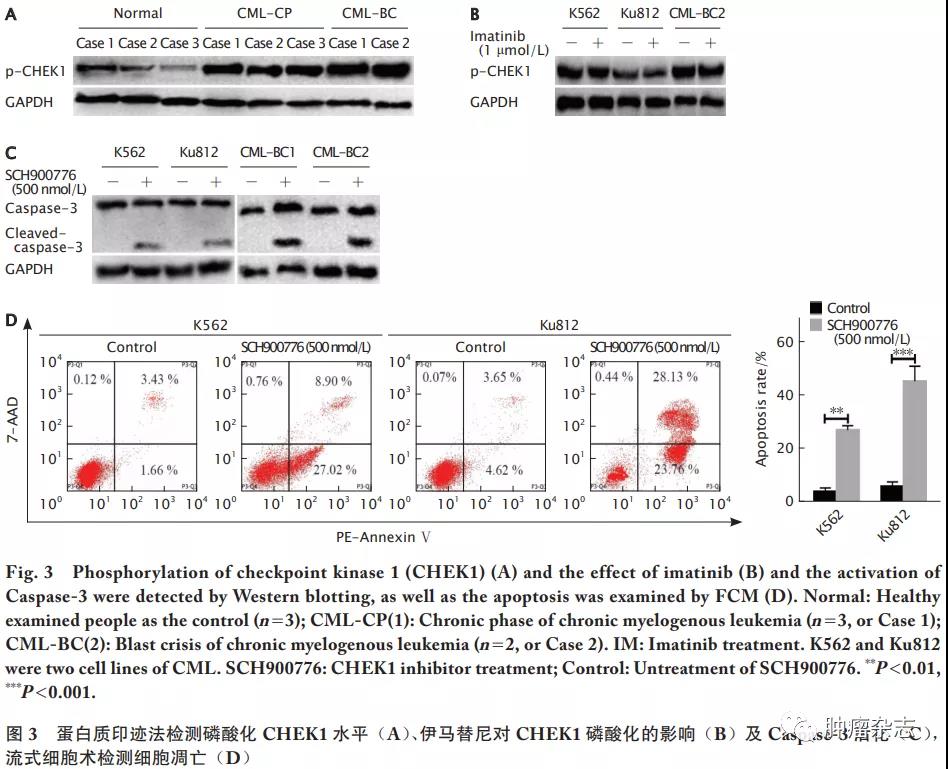
相比正常体检者,CML患者外周血单个核细胞中CHEK1 mRNA水平明显升高,慢性期患者约上调2倍(P<0.05),急变期进一步上调,约为正常组的3.8倍(P<0.01)(图1A)。在蛋白水平上,CHEK1表达也呈现逐步增加的趋势,其中正常体检对照组的CHEK1相对表达丰度均值为0.018,而慢性期患者的均值约为0.62,急变期则为1.12(图2B)。K562、Ku812和来源于急性期CML患者的单个核细胞经伊马替尼(1μmol/L)处理后,与未处理对照组相比,CHEK1蛋白的表达水平均无明显差异(图2C)。


2.3
抑制CHEK1磷酸化促进CML细胞凋亡
CHEK1磷酸化水平与总CHEK1表达具有相同的变化趋势,其相对表达丰度均值由正常体检者的0.42增加到慢性期CML的1.24,急变期CML增加到2.03(图3A);而且CHEK1磷酸化水平并未受伊马替尼处理影响(图3B)。CHEK1特异性抑制剂SCH900776能够有效降低CHEK1磷酸化水平,且活化半胱天冬酶-3(图3C),同时有效诱导细胞凋亡,K562和Ku812细胞的凋亡率分别为23.5%(P<0.01)和42.7%(P<0.001)(图3D)。
2.4
抑制CHEK1磷酸化增加G2/M期细胞所占百分比
SCH900776可明显改变细胞周期分布,加速S期向G2/M期转化;K562和Ku812细胞中,S期所占百分比分别由(28.3±3.44)%和(25.5±2.97)%降低至(15.6±5.86)%和(12.8±4.86)%(P 值均<0.05),同时G2/M期所占百分比由(42.1±2.99)%和(32.3±4.46)%增加到(59.4±5.30)%和(48.5±6.04)%(P 值均<0.05)(图4A)。在8例临床患者的外周血单个核细胞中,除2例没有明显变化外,其余皆可观察到S期细胞显著减少(图4B)。用特异性抑制剂SCH900776抑制CHEK1磷酸化后,下游靶蛋白CDC25A表达水平上调,p-CDK2蛋白表达水平降低(图4C)。

2.5
抑制CHEK1磷酸化后DNA损伤修复受阻
&γ;-H2AX作为DNA损伤标志物,是DNA双链断裂后修复的最早期事件之一。K562和Ku812细胞及外周血标本来源的单个核细胞,用特异性抑制剂SCH900776处理后可抑制CHEK1磷酸化且均能引起&γ;-H2AX表达水平的升高(P<0.05)。随后分析了p53家族3个成员p53、p63及p73的表达水平,结果(图5)显示,p53由于在K562和Ku812细胞中均缺失,未能检测到表达,而在急变期CML第2例样本中p53表达水平明显升高(P<0.05);p63在3组处理对象中表达没有明显变化;p73则在细胞株及标本中均有明显升高(P<0.05,P<0.01)。

3 讨 论
细胞在其生理过程中面临着各种DNA损伤压力,外源性损伤如紫外线、辐射和化学药物等刺激,内源性损伤来源于细胞分裂过程中DNA复制错误积累[9]。DNA损伤修复是决定细胞命运的关键。尤其在肿瘤细胞中,失控的增殖需要更为旺盛的代谢能力,而代谢过程所产生的氧自由基等物质是造成DNA损伤的重要成分[10]。因此,肿瘤细胞也进化出了相对强大的DNA修复系统[11]。
DNA损伤修复主要有ATM-CHEK2和ATR-CHEK1两条信号通路执行[12]。目前研究认为,前者主要介导复制叉停滞(replication fork),引起局部修复行为;后者主要调节复制起始,激活后能够全局抑制DNA复制。在细胞分裂的S期,ATM和ATR作为损伤感应蛋白,能够快速识别损伤点,并磷酸化活化CHK2和CHEK1,CHEK1/2再进一步磷酸化CDC25A,促进其向细胞质转运后降解。CDC25A降解导致其靶蛋白CDK2-Cyclin E复合物由于持续磷酸化而失活,下游复制起始蛋白CDC45不能有效结合DNA,形成限制点,最终复制停止,细胞周期阻滞在S期[13]。S期限制点延缓DNA复制,为细胞进行修复提供了时间。因此,DNA损伤修复是一道重要的自我保护机制。若损伤过于严重或持续存在,修复机制失效,细胞则启动自杀程序,以避免因损伤造成的染色体异常传递到子细胞中[14]。
作为DNA修复反应的重要调节分子,CHEK1在肿瘤细胞中过表达,提示肿瘤细胞更加依赖其缓解复制过程中产生的损伤应激[15]。因此可以作为攻击肿瘤细胞的靶点。本研究则以CML细胞为对象,证实CHEK1 mRNA及蛋白在CML细胞中表达水平升高,抑制其激酶活性能明显诱导肿瘤细胞凋亡。其次,在细胞周期分布上,S期细胞所占百分比明显降低,提示CHEK1只有作为引起S期阻滞的重要功能。CHEK1在CML细胞中表达上调,可能是肿瘤细胞中含有相对较多内源性DNA损伤的补偿和适应结果。有研究显示,CHEK1抑制后细胞凋亡发生在G2/M期,这可能与抑制CHEK1后破坏S期限制点,DNA损伤得不到及时修复便进入G2/M期,并启动凋亡程序有关[16]。本研究中损伤标志物&γ;-H2AX持续存在,也证明了这一点。同时在分子水平,本研究发现抑制CHEK1后CDC25A磷酸化水平降低,CDC25A能够去磷酸化激活CDK2;另外,CDC25A磷酸化降解减少,总CDC25A水平升高,相应地,CDK2磷酸化水平降低而活性增加,DNA复制持续进行。因此,抑制CHEK1后CDC25A及CDK2磷酸化水平降低,提示S期限制点被破坏[17]。
CHEK1抑制后细胞凋亡依赖半胱天冬酶途径,半胱天冬酶途径激活主要依赖转录因子p53对Bcl-2家族促凋亡蛋白的表达调节。而在K562和Ku812细胞中,p53基因缺失突变,半胱天冬酶活化可能不依赖于p53蛋白,但p73显著升高,推测CHEK1可能是细胞凋亡相关的主要分子[18]。有研究发现,DNA损伤感应蛋白ATM能够同时磷酸化活化C-Jun氨基末端激酶(C-JunN-terminal kinase,JNK)和p38等,后者可不依赖P53而上调Bcl-2家族促凋亡蛋白BIM,从而激活线粒体凋亡途径。因此,后续研究抑制CHEK1诱导细胞凋亡的分子机制可聚焦于JNK-BIM信号轴[19]。
综上所述,本研究证实了CHEK1在CML细胞株及临床样本中处于过表达状态,抑制CHEK1磷酸化可破坏S期DNA修复,导致DNA损伤积累,从而诱导细胞凋亡。另外本研究还发现,CHEK1上调表达并不受BCR/ABL活化状态的影响,这在TKIs耐药研究中具有更重要的意义。
更多《肿瘤》杂志文章,请访问以下网址:
https://navi.cnki.net/knavi/JournalDetail?pcode=CJFD&pykm=ZZLL
参考文献
[1]Maru Y. Molecular biology of chronic myeloid leukemia[J]. Cancer Sci, 2012, 103(9):1601-1610.
[2]Capdeville R, Buchdunger E, Zimmermann J, et al. Glivec(STI571, imatinib), a rationally developed, targeted anticancer drug[J]. Nat Rev Drug Discov, 2002, 1(7):493-502.
[3]Corbin AS, LA Rosee P, Stoffregen E, et al. Several Bcr-Abl kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib[J]. Blood, 2003,101(11):4611-4614.
[4]Griswold I, Macpartlin M, Bumm T, et al. Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib[J]. Mol Cell Biol, 2006, 26(16):6082-6093.
[5]Krishna R, Bubnoff N, Miething C, et al. Imatinib and leptomycin B are effective in overcoming imatinib-resistance due to Bcr-Abl amplification and clonal evolution but not due to Bcr-Abl kinase domain mutation[J]. Haematologica, 2008, 93(11):1718-1722.
[6]Cornelison M, Kantarjian H, Cortes J, et al. Outcome of treatment of chronic myeloid leukemia with second-generation tyrosine kinase inhibitors after imatinib failure[J]. Clin Lymphoma Myeloma Leuk, 2011, 11(Suppl 1):S101-110.
[7]Hochhaus A, La Rosée P. Imatinib therapy in chronic myelogenous leukemia: strategies to avoid and overcome resistance[J]. Leukemia, 2004, 18(8):1321-1331.
[8]Yang K, Fu LW. Mechanisms of resistance to BCR-ABL TKIs and the therapeutic strategies: A review[J]. Crit Rev Oncol Hematol, 2015, 93(3):277-292.
[9]Rodemann P, Dittmann K, Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair[J]. Int J Radiat Biol, 2007, 83(11/12):781-791.
[10]Hakem R. DNA-damage repair; the good, the bad, and the ugly[J]. EMBO J, 2008, 27(4):589-605.
[11]Cohn D, Frankel W, Resnick K, et al. Improved survival with an intact DNA mismatch repair system in endometrial cancer[J]. Obstet Gynecol, 2006, 108(5):1208-1215.
[12]Bartek J, Lukas J. CHK1 and CHK2 kinases in checkpoint control and cancer[J]. Cancer Cell, 2003, 3(5):421-429.
[13]Smith J, Tho LM, Xu NH, et al. The ATM-CHK2 and ATR-CHK1 pathways in DNA damage signaling and cancer[J]. Adv Cancer Res, 2010, 108:73-112.
[14]MICHELENA J, GATTI M, TELONI F, et al. Basal CHK1 activity safeguards its stability to maintain intrinsic S-phase checkpoint functions[J]. J Cell Biol, 2019, 218(9):2865-2875.
[15]Michelena J, Gatti M, Teloni F, et al. Targeting CHK1 inhibits cell proliferation in FLT3-ITD positive acute myeloid leukemia[J]. Leuk Res, 2014, 38(11):1342-1349.
[16]Capasso H, Palermo C, Wan SH, et al. Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest[J]. J Cell Sci, 2002, 115(Pt 23):4555-4564.
[17]Zhou XY, Wang X, Hu BC, et al. An ATM-independent S-phase checkpoint response involves CHK1 pathway[J]. Cancer Res, 2002, 62(6):1598-1603.
[18]Wakatsuki M, Ohno T, Iwakawa M, et al. p73 protein expression correlates with radiation-induced apoptosis in the lack of p53 response to radiation therapy for cervical cancer[J]. Int J Radiat Oncol Biol Phys, 2008, 70(4):1189-1194.
[19]Llopis A, Salvador N, Ercilla A, et al. The stress-activated protein kinases p38α/β and JNK1/2 cooperate with CHK1 to inhibit mitotic entry upon DNA replication arrest[J]. Cell Cycle, 2012, 11(19):3627-3637.
